
More Information
Submitted: March 09, 2021 | Approved: March 24, 2021 | Published: March 25, 2021
How to cite this article: Guerrier D, Morcel K. Partial SHOX duplications associated with various cases of congenital uterovaginal aplasia (MRKH syndrome): A tangible evidence but a puzzling mechanism. J Genet Med Gene Ther. 2021; 4: 001-008.
DOI: 10.29328/journal.jgmgt.1001006
Copyright License: © 2021 Guerrier D, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: CNV; Congenital uterovaginal aplasia; Duplication; MRKH syndrome; SHOX

Partial SHOX duplications associated with various cases of congenital uterovaginal aplasia (MRKH syndrome): A tangible evidence but a puzzling mechanism
Daniel Guerrier1* and Karine Morcel1,2
1University Rennes, CNRS, IGDR (Institut de Génétique et Développement de Rennes), UMR 6290, Rennes, France
2Hospital Center Regional University Morvan De Brest, Brest, France
*Address for Correspondence: Daniel Guerrier, Rennes 1 University, IGDR – CNRS UMR, 6290, Villejean Medical Campus, 35043, Rennes, France, Tel: +33 (0)223 234 679; Email: [email protected]
The Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome is the most severe form of congenital malformation of the inner female reproductive tract. It is diagnosed as such when the uterus, the upper vagina and optionally the Fallopian tubes are absent. It accounts for approximately 1 in 5000 live-born females and has been classified in two subtypes: type 1 in the presence of isolated uterovaginal aplasia and type 2 when associated in various combinations with extragenital malformations of the kidneys, skeleton, heart and auditory system. Most cases of MRKH syndrome are sporadic, although a significant number of many familial cases have been reported to date. Despite numerous studies, the genetics of the syndrome remains largely unknown and appears to be heterogeneous: chromosomal abnormalities and some candidate gene variants appear to be associated with a few cases; others have been suggested but not yet confirmed. To date, mainly the GREB1L gene appears to be a serious candidate. Among the remaining hypotheses, the controversial contribution of partial duplications of the SHOX gene is still puzzling, as the deficiency of this gene is a major cause of skeletal adysplasia syndromes. We have attempted to resolve this controversy in a study of 60 MRKH cases. Our results tend to show that SHOX duplications can be the origin of a genetic mechanism responsible for MRKH syndrome.
Among congenital malformations of the female genital tract (FGT), aplasia of the uterus and vagina, generally referred to as Mayer-Rokitansky-Küster-Hauser syndrome (MRKHS) (OMIM #277000), is the most severe form [1,2] with an incidence estimated to about 1 in 5000 newborn females [3]. However this syndrome is not as easy to diagnose as such in the first instance, since the aplasia of the uterus may be total or partial, that of the vagina is of various length, and Fallopian tubes affection ranges from total aplasia to almost complete structures. Attempts to classify FGT malformations illustrate the difficulties to reach a consensus [4,5,6,7]. The only common feature appears to be a primary amenorrhea in phenotypically female subjects (normal development of secondary sexual characteristics [1] and normal external genitalia), indicating the presence of functional ovaries with no sign of hyperandrogenism [8], in an otherwise normal XX chromosomal background. At this first stage, a differential diagnosis can already be undertaken to avoid an initial wrong direction [1]. The syndrome is defined as MRKH type 1 when a complete aplasia of the uterus and vagina is observed without any associated malformations, but where Fallopian tubes are most often present. On the other hand, MRKH type 2, includes hypoplasic to aplastic uterus, upper vagina aplasia and variable affection of Fallopian tubes, the whole being associated with a wide range of malformations, themselves found with various degrees of frequency [9,10,11]. They are mainly renal (unilateral agenesis, ectopic or horseshoe kidney), skeletal (scoliosis, Klippel-Feil anomalies, hemivertebrae) and to a lesser extend auditory or cardiac. During this last decade, thorough analysis of large cohorts, has allowed to gather consistent data on the nature and frequency of associated malformations. In spite of some yet unexplained geographic variations [10,12], isolated uterovaginal aplasia (MRKH type 1) and association of renal or skeletal malformations (MRKH type 2) remain the most frequent forms encountered worldwide with around 60% of type 1 and 40% of type 2 [11].
Once the genetic origin of the syndrome was admitted [13,14,15], the approaches to find the cause have followed technological developments to currently lead to whole exome/genome sequencing (WES/WGS), via hypothesis-based mutational analysis of candidate genes, comparative genomic hybridization (CGH) array, or the search for deletions or duplications of targeted chromosomal regions by multiplex ligation-dependent PCR assay (MLPA) [16]. To the best of our knowledge, none of the hypothesized candidate genes could formally account for the syndrome [9,11]. Numerous chromosome imbalances, mainly segmental deletions, have been associated with MRKHS using CGH array, but their pathogenicity has not been established for the vast majority of them [9]. However two recurrent genomic rearrangements mapping to 17q12 [17,18,19] and 16p11.2 [18] were found in several unrelated cases of MRKHS of both types. These two loci respectively include the LHX1 and TBX6 genes which were considered as strong candidates based on their documented role during development of the female genital tract. Subsequent mutational analysis of both genes evidenced this role, but pathogenic variants were found in a very limited number of cases, in LHX1 [20,21,22,23], and in TBX6 [22,24]. Nowadays, the third generation sequencing allows very fine linkage analysis through WGS and sequencing of (almost) all coding regions of the genome (WES). These technologies have recently been applied to the genetics of MRKHS, first on a selected group of MRKH type 1 patients, which led to unveil new candidate genes (PIK3CD, SLC4A10 and TNK2) bearing putatively pathogenic variants [25]. Two other teams using a WES approach, identified different GREB1L gene variants from a total of 5 families and 6 sporadic cases [26,27]. Data from these latter studies also suggest that GREB1L is the first major gene involved in MRKHS. Indeed its involvement in familial cases of uterovaginal aplasia associated with renal adysplasia, together with other sporadic cases of isolated uterovaginal aplasia (type 1) or associated with skeletal or facial malformations (type 2), reinforces this assumption. This also suggests that, beyond its involvement in kidney [28] and probably female genital tract development [29], GREB1L could play a more pleiotropic role during development and be involved in other malformative syndromes.
Despite the discovery of this new gene, there are still many unexplained cases of MRKHS for which genetic studies have not been completely successful. This applies to the various chromosomal rearrangements involving genes not yet characterized as well as to the controversial association of various partial heterozygous duplications of the SHOX gene with MRKHS. Indeed, non-overlapping duplications of this gene were described using the MLPA technique, in cases of MRKHS type 1, familial (father-transmitted) or sporadic [30]. This study was later contradicted through analysis of a larger cohort by mean of the same technique [31], but this result remains puzzling, especially considering the relatively high frequency of these duplications within the MRKH cohort of patients analyzed (5/30) [30]. It is even more puzzling that the only role assigned to the SHOX gene, is its involvement in some skeletal dysplasia syndromes such as Léri-Weill dyschondrosteosis (LWD, MIM 127300), Langer mesomelic dysplasia (LMD, MIM 249700), idiopathic short stature (ISS, MIM 300582) or its contribution to Turner syndrome, all attributable to various deletions, duplications or mutations within the coding or regulatory sequences of the SHOX gene [32,33,34]. This is why we wanted to contribute to this debate by studying a cohort of 60 patients using the same MLPA technique. We found four duplications of various lengths, within or adjacent to the SHOX gene, some similar to those described previously, and some new ones, in four sporadic cases. The association of these genetic events with MRKHS seems then relevant and we discuss it in technical and mechanistic terms.
Patients
We studied a cohort of 60 women who had utero-vaginal aplasia diagnosed by clinical examination and transabdominal ultrasonography and/or magnetic resonance imaging (MRI) or celioscopy. All patients had a normal karyotype 46, XX. They were examined for associated malformations using renal ultrasound, spinal X-ray, echocardiogram and audiogram, as necessary. Twenty (~33%) had isolated utero-vaginal aplasia (MRKH type I). The remaining women (~67%) had a type 2 syndrome with renal, spinal or other skeletal malformations (including Klippel-Feil sequence, Sprengel deformity, digit defects such as clinodactyly, brachydactyly or syndactyly), cardiac anomalies or hearing impairment. All the subjects were enrolled through a French national multicentric research program, called PRAM (Programme de Recherche sur les Aplasies Müllériennes). This study has been approved by the local institutional review board (Project # 05/16-543), and is registered with the French Ministry of Health (DGS # 2005/030). Informed consent was obtained from all subjects.
The four patients in whom we found partial duplications of the SHOX gene, showed MRKH syndrome type 2 (Table 1). There were no other affected siblings, nor was there any family history to report.
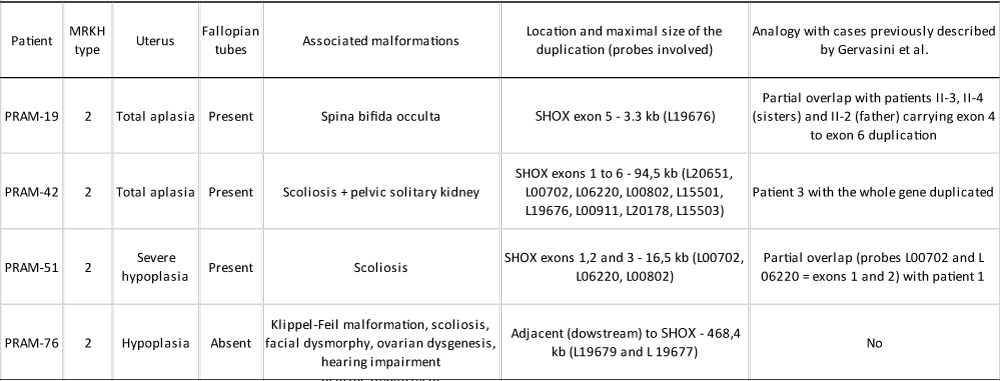
Table 1: Diagnosis and main clinical features of patients with MRKH syndrome heterozygous for partial SHOX gene duplications. Genetic similarities with the cases previously described by Gervasini and collaborators [30]. All patients showed normal size, weight and BMI for their age.
Methods
Genomic DNA was extracted from whole blood of patients or healthy control subjects, using the QIAamp DNAKit (Qiagen, Venlo, Netherlands) according to the manufacturer’s protocol.
MLPA analysis was performed using the commercial SALSA MLPA kit PO18-F1 SHOX (MRC-Holland, Amsterdam, Netherlands) in accordance with the manufacturer’s instructions. P018 SHOX probe mix contains probes for each exon of the SHOX gene and some other targeting upstream and downstream regions of the gene, meant to be involved in its transcriptional regulation. More precisely, this kit includes 48 probes, six of which target the exons 1 to 6, two the intron 6, thirteen its regulatory or flanking regions and seven target other genes within the pseudoautosomal region 1 (PAR1) of the X and Y chromosomes where the SHOX gene locates. Eight other probes serve as internal control for the X chromosome outside the PAR1 region and 12 are external to this chromosome, of which 2 are specific for the Y chromosome. Finally, two extra probes provide amplification and denaturation controls. MLPA reactions were achieved from 200 ng of genomic DNA and PCR products were afterwards subjected to capillary electrophoresis using an ABIPRISM 3100 genetic analyzer (Applied Biosystems, Foster City, CA, USA). Electrophoretic patterns were analyzed using the GeneMarker software (Softgenetics, State College, PA, USA) which uses the internal control probes to normalize peak heights and then compares tests samples to a normal control panel. This latter consists in five normal genomic DNAs which are individually MLPA-processed and for which the peak areas of each amplification are averaged and then compared to each peak area of the DNA of the patient under study. Each patient was thus analyzed at least twice independently.
SHOX duplications were confirmed by another semi-quantitative technique using the multiplex PCR/liquid chromatography (MP/LC) method [35] in a duplex assay (DP/LC) that we had already proven in two previous studies [36,37]. Briefly, two target genomic sequences, from an internal control HMBS gene (hydroxymethylbilane synthase gene located at 11q23.2-qter) and from the region found duplicated, were amplified using unlabeled specific primers in the same PCR reaction. Primers (Table 2) were designed using the Primer Premier Software (Premier Biosoft International, Palo Alto, CA, USA) and in order to amplify genomic sequences overlapping or adjacent to those targeted by the MLPA probes. Experiments were achieved in parallel from a pool of five genomic DNAs of control subjects and from DNA of a patient of interest. PCR products were then separated by high-performance liquid chromatography (HPLC), and quantitated by fluorescent detection using a post-column intercalation dye. Navigator™ Software (Transgenomic, Omaha, NE, USA) was used for data analysis and the HMBS peak was used for normalization; relative peak intensity for each SHOX amplicon directly reflected genomic copy number.
| Table 2: Gene location and sequences of primers used in the DP/LC experiments. Different combinations of HMBS/SHOX primers were used according to the best compatibility calculated by the Primer Premier 5 software which also provides the best ΔG (kcal/mol) value for each oligonucleotide in a given mix of HMBS/SHOX sens and antisens primers. Consequently, the HMBS exon 14 primer pair was used in combination with 3 different SHOX primer pairs (upper part of the table) and the HMBS exon 15 primer pair with 4 different other SHOX primer pairs (lower part of the table). | ||||
| Location | Sens primer (5’-3’) | Antisens primer (5’-3’) | PCR size | |
| Exon 14 | TAGACGGCTCAGATAGCATACAAG | AGGTGGGATTTGGTGAGAACA | 132 nt | |
| 4 kb upstream | CGGCACAAATTAGGCCATTA | CATTGCCTGTCGGGTGAAA | 147 nt | |
| Exon 5 | GAGCCTTACGGATGCCTTTC | CAGGATGCGGCAGCAAAT | 107 nt | |
| Intron 6 | GAGAAGCCGGTTAAGGAATGTA | CGGGACCTGCACGTACAAT | 113 nt | |
| Exon 15 | AGACCATGCAGGCTACCATCC | GGTCATCCTCAGGGCCATCT | 148 nt | |
| Exon 1 | GGTGCAAAGGCGAGGAGA | AATGCCCAGGGTGCTGAC | 166 nt | |
| Exon 2 | TCCAGGACATCACGGAGGG | CCGGAGCGCAAAGGAACT | 132 nt | |
| Exon 3 | GCGTCAAAGCGCATTGGT | TCCTCGTCCTCCGACTTCA | 170 nt | |
| Exon 6 | CGCCCTACCTGATGTTCCC | GGTCGGCGATGCTGGAAT | 129 nt | |
| Exon 7 | TCCTGGGCTCAAGCAATCC | CCTCAGCAGCAAAGCAAGATC | 124 nt | |
Partial heterozygous duplications within or in the vicinity of the SHOX gene were once described in association with the MRKH syndrome in four independent cases (3 isolated and 2 sisters with the same phenotype). These results were obtained through a genetic analysis by MLPA and were confirmed by haplotyping and pyrosequencing [30]. However, they were challenged by the study of a larger MRKH cohort where no relevant duplication of SHOX was found using the same commercial SALSA MLPA kit PO18-E1 SHOX [31]. Since then, no further studies have been reported to try to resolve this controversy, except for the recent description of a new case of MRKH showing a heterozygous duplication of a distant regulatory element of the SHOX gene [38]. In the present study, we analyzed a cohort of 60 MRKH patients and found various SHOX duplications in four independent patients (Figure 1). These duplications involved different exons of the SHOX gene for three of them and a region close to the gene for the fourth (Table 1). The duplications were also confirmed by another gene dosage technique, DP/LC (Figure 2). These results raise then several questions: 1) are duplications found in MRKH patients significant and thus, can they be associated with the syndrome? 2) If so, can this syndrome be attributable to defects in the SHOX gene? 3) If not, what is the genetic link between these duplications and MRKH syndrome?

Figure 1: SHOX gene dosage in four independent MRKH patients (PRAM-16, -19, -42 and -51) assessed by the MLPA kit PO18-F1 SHOX. In the diagrams, MLPA probes are represented along the x-axis (size of PCR products) and the fluorescent intensity ratio is represented on the y-axis. Each probe is represented by a square (green for SHOX gene and surrounding regions, blue for internal controls, grey for the Y chromosome). The correspondence between the probes size and their respective location is shown on Supplementary Data S1. The upper and lower arbitrary borders are shown respectively as a green upper and lower line. Probe ratios crossing the upper or the lower border are respectively indicative for a duplication or a deletion. Thus, a ratio of 1.5 (3:2) indicates the presence of an additional copy (heterozygous duplication) of a DNA stretch of a gene present in two copies in the genome, while a ratio of 0.5 (1:2) would indicate a heterozygous deletion. The detailed analysis of MLPA experiments is also included in Supplementary DataS1. (A) Results of patient PRAM-16 showing no copy number variations. (B) Results of patient PRAM-19 showing a heterozygous duplication of 1 probed region (SHOX exon 5). (C) Results of patient PRAM-42 showing the duplication of 9 contiguous probed regions (4.7 kb upstream of the SHOX gene up to end of intron 6, 1.4 kb upstream of exon 7) and (D) Results of patient PRAM-51 showing the duplication of 3 contiguous probed regions (SHOX exons 1, 2 and 3).
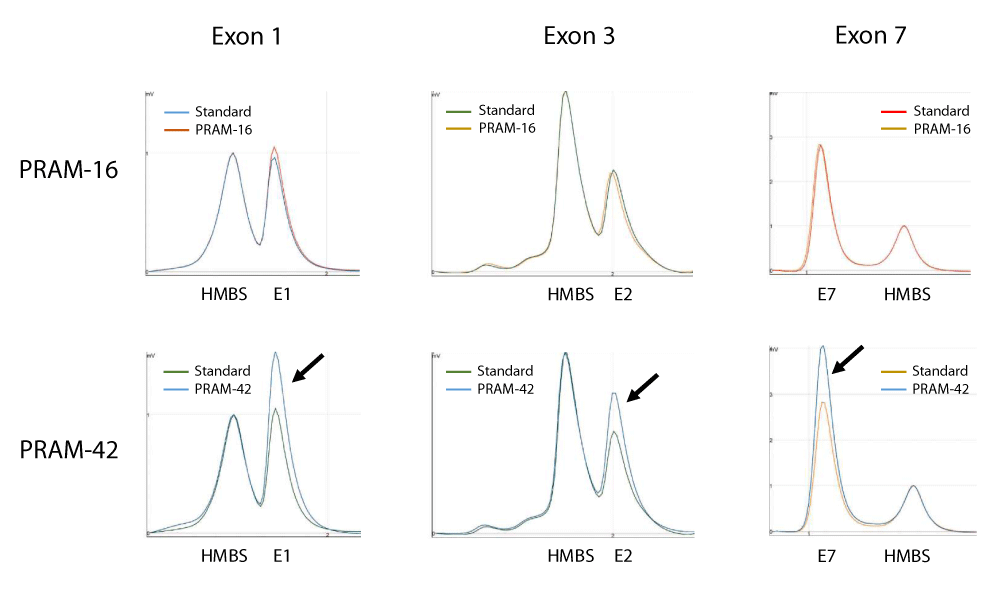
Figure 2: DP/LC chromatograms for three different duplexes used in SHOX analysis. x-axis: retention time in min; y-axis: fluorescence intensity. Example of results obtained for quantification of copy number of SHOX exons 1, 3 and 7 in patients PRAM-16 and PRAM-42. In these experiments, a pool of 5 independent genomic DNAs was used as standard and the hydroxymethylbilane synthase (HMBS) gene, located at 11q23.2-qter, was used as an internal control. Profiles are superimposed and then normalized using the control amplicon for HMBS. Black arrows show the triple dosage of SHOX exons 1, 3 and 7 in patient PRAM-42. Primers sequences and location are summarized in Table 2.
Significance of SHOX duplications and association with MRKH syndrome.
First, it seems that the heterozygous SHOX duplications found by MLPA are not experimental artifacts as they were corroborated by another method, DP/LC, allowing to measure the copy number of genomic sequences overlapping or adjacent to those targeted by the MLPA probes. Moreover, the fact that duplications of contiguous regions were found when, in the technique, there is no collinearity between the size of the corresponding PCR products and their position on the gene (typically the case for SHOX exon probes), attests to the absence of artifacts. Are these duplications then simple copy number variants (CNV) or are they associated with MRKH syndrome? In the work of Gervasini and colleagues [30], in ours as well as in other investigations using the same commercial MLPA kit [33,39,40], generally no CNV or at least no likely pathogenic variants were found in the control DNAs, whereas Sandbacka and colleagues [31] found as many in each patient and control group. In this latter study, the so-called genomic aberrations (duplications but also deletions) often concerned the same SHOX areas (downstream of the gene) in both groups, which is rather suggestive of a defective standardization. In addition, it appears that no alternative technique was used to verify the copy number of the regions found aberrant. Finally the use of genomic DNAs prepared with two very remote techniques (Puregene DNA Isolation kit and phenol-chloroform method) despite the instructions from the MPLA kit supplier, also may provoke artifacts. This seems all the more plausible since the variants described by Sandbacka and colleagues [31] are listed in the Database of Genomic Variants (DGV) while those described by Gervasini and colleagues [30] as well as those of our present study, are not and involve inner parts of the gene, reflecting probably an underlying relationship with MRKH syndrome.
Can SHOX duplications be responsible for MRKH syndrome?
The SHOX (short stature homeobox-containing gene on chromosome X) gene locates on the pseudoautosomal region (PAR1) on the short-arm tips of both X (Xp22.33) and Y (Yp11.3) chromosomes and escapes X-inactivation [41]. It encodes a transcription factor which plays a pivotal role in bone elongation and two copies of the gene are required for normal skeletal development [42]. It is the major gene involved in different skeletal dysplasia syndromes such as Léri-Weill dyschondrosteosis (LWD), Langer mesomelic dysplasia (LMD), idiopathic short stature (ISS), and it contributes to the phenotypic manifestations of Turner syndrome. Intragenic SHOX deletions and duplications of various size, mutations or deletions within the SHOX coding or regulatory sequences, or monosomy X0 account for the skeletal defects in these syndromes [32,34,43,44]. By contrast, SHOX overdosage either by experimental overexpression [45] or due to structural or numerical abnormalities of the sex chromosomes (mainly X trisomy), can lead to long limbs and tall stature [42]. When limited to the entire SHOX gene and neighboring (regulatory) sequences, the associated phenotype is also restricted to height, with normal or tall stature [46,47,48]. Surprisingly, about as many partial deletions as partial duplications have been reported in large-scale studies of subjects with skeletal dysplasia syndrome [49,44]. Furthermore, partial SHOX duplications, as well as those encompassing SHOX transcription enhancers, have appeared to be highly penetrant alleles in respect to LWD/ISS [40,49,50] and have more deleterious effects in regards to skeletal dysplasia and height gain, than complete SHOX duplications. However, while partial SHOX deletions have always been associated with skeletal defects [33,40,49], partial SHOX duplications have been identified in patients with variable and non-overlapping phenotypes, as different as LWD and ISS, autism spectrum disorders and related neurodevelopmental conditions [51], or MRKH syndrome [30], as already pointed out [44]. The hypothesis that the variable clinical manifestations associated with partial or total duplications of SHOX are probably dependent on the physical location of the duplicated sequence [40,44], then seems the most plausible.
What could be the genetic/mechanistic link between partial SHOX duplications and MRKH syndrome?
This question is all the more relevant since there is occasional overlap between the numerous duplications associated with LWD/ISS and those identified in MRKH patients, even though these two types of syndromes do not have common phenotypic manifestations. It is in the answer to this question that the hypothesis stated above takes on its full meaning. Indeed, haploinsufficiency of the SHOX gene, when mutated, partially or totally deleted, or deprived of its regulatory sequences, is a major cause of skeletal dysplasia [34]. The numerous cases of LWD/ISS associated with partial heterozygous duplications of SHOX can then only be explained by the intragenic insertion of the duplicated sequences interrupting the reading frame of the gene or interfering with its transcriptional regulation. This pathogenic mechanism was already suggested through the cytogenetic localization of the duplicated SHOX sequences in patients with LWD or ISS [40,49] or by DNA sequencing at the fusion junction of the duplication showing integration of the duplicated sequence at a genomic region adjacent to the original position [49]. On the other hand, SHOX duplications, when associated with MRKHS, cannot have been inserted within the sequences of SHOX without manifesting as LWD or ISS. This proposal is furthermore supported by the absence of the phenotypic characteristics of LWD or ISS syndromes in the biggest cohorts of MRKH patients previously studied worldwide [3,9,10,23,52,53] and by the absence of uterine malformations in LWD or ISS patients [34,54]. This raises the question of whether similar duplications can be inserted either in the vicinity of their homologous sequences (intrachromosomal duplication) or within another chromosome (interchromosomal duplication). During meiosis and especially in males, recombination between highly similar duplicated sequences (non-allelic homologous recombination) can occur and generate deletions, duplications, inversions and translocations throughout the genome, and it is responsible for genetic diseases known as ‘genomic disorders’, most of which are caused by altered copy number of dosage-sensitive genes [55]. This is particularly the case for the obligatory exchanges that occur between Xp/Yp pseudoautosomal regions (PAR1). Crossovers in this 2.6 Mb chromosomal segment, create a male-specific recombination ‘hot domain’ with a recombination rate that is about 20 times higher than the genome average [56]. More specifically, the 231 kb interval encompassing the SHOX gene, shows a 27-fold increase recombination relative to the genome average rate [57]. This characteristic may be due to the presence of numerous repeated sequences, Alu-like elements or rich A-T regions identified at certain breakpoints, such as in intron 3 [44] or downstream of the SHOX gene [33]. These various sequences are then likely to promote non-allelic homologous recombination by unequal intra- or inter-chromosomal crossing-over, thus generating deletions and duplications (nonrecurrent CNVs) [58] underlying LWD/ISS and MRKHS. The relative small size of microdeletions (~3 to 374 kb) and microduplications (~ 3 to 571 kb) identified in both LWD/ISS and MRKHS in previous studies [30,40,44] and in the present one (Table 1), seems to reflect the high density of recombinogenic sequences found in the PAR1 region and even more in the interval comprising the SHOX gene. Moreover, it appears that multiple combinations are possible to generate deletions or duplications from these active sequences, some being used predominantly [33,39,44,49,59]. Various genetic outcomes for duplication CNVs have been demonstrated, including the pathogenicity of some intergenic duplications [60]. The insertion of duplicated SHOX sequences into another locus, resulting in the disruption of the reading frame and the loss of function of a host gene, is therefore the sole explanation for associating MRKH syndrome with these duplications. Thus the inactivation of a dosage-sensitive host gene could take place while keeping intact the original template SHOX gene at its locus. The identification of such host gene by next-generation approaches [60], would certainly lead to the characterization of new genetic cause(s) for MRKHS.
To conclude, the absence of phenotypic overlap between LWD/ISS and MRKHS suggests, in the case of SHOX duplications, different mechanisms of generation and insertion of these duplications, the study of which could be beneficial to medical genetics. The data obtained in this study tend to demonstrate that the SHOX gene is not involved in MRKH syndrome and encourages future investigations to identify the gene(s) that the insertion of SHOX duplications involve in this syndrome.
We first wish to thank the members of both MAIA (https://maia-asso.org) and Syndrome de Rokitansky-MRKH (https://asso-mrkh.org) associations for their contribution, and particularly, the patients with MRKH syndrome who participated in our research program. We are also indebted to all physicians and researchers involved in the French national PRAM (Programme de Recherches sur les Aplasies Müllériennes) network, and to Dr. Anne-Marie Jouanolle, who provided us with DNA samples from healthy volunteer donors. We also want to acknowledge the involvement of short-term trainees who participated in this research program: Pauline Berneau, Solène Duros, Lydia Flaux, Pauline Sararols and Galaad Schorp. We are finally grateful to Dr. Christèle Dubourg for assistance and helpful discussions during technical settings of DP/LC, and to Stéphane Dréano for the development of the analysis of MPLA products by capillary electrophoresis, for taking in charge our samples as well as for his availability and kindness. This work was not supported by any particular grant other than the annual recurrent budget of the CNRS for the team. D. Guerrier is a tenured researcher at the “Institut National de la Recherche Médicale“ (INSERM).
- Morcel K, Camborieux L, Guerrier D. Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome. Orphanet J Rare Dis. 2007; 14: 13. https://ojrd.biomedcentral.com/articles/10.1186/1750-1172-2-13
- Jacquinet A, Millar D, Lehman A. Etiologies of uterine malformations. Am J Med Genet A. 2016; 170: 2141-2172. https://onlinelibrary.wiley.com/doi/abs/10.1002/ajmg.a.37775
- Herlin M, Bjørn AM, Rasmussen M, et al. Prevalence and patient characteristics of Mayer-Rokitansky-Küster-Hauser syndrome: a nationwide registry-based study. Hum Reprod. 2016; 31: 2384-2390. https://academic.oup.com/humrep/article/31/10/2384/2198191
- Acién P, Acién M, Sánchez-Ferrer M. Complex malformations of the female genital tract. New types and revision of classification. Hum Reprod. 2004; 19: 2377-2384. https://academic.oup.com/humrep/article/19/10/2377/589016
- Oppelt P, Renner SP, Brucker S, et al. The VCUAM (Vagina Cervix Uterus Adnex-associated Malformation) classification: a new classification for genital malformations. Fertil Steril. 2005; 84: 1493-1467. https://www.fertstert.org/article/S0015-0282(05)02786-X/fulltext
- Acién P, Acién MI. The history of female genital tract malformation classifications and proposal of an updated system. Hum Reprod Update. 2011; 17: 693-705. https://academic.oup.com/humupd/article/17/5/693/759275
- Grimbizis GF, Gordts S, Di Spiezio Sardo A, et al. The ESHRE/ESGE consensus on the classification of female genital tract congenital anomalies. Hum Reprod. 2013; 28: 2032-2044. https://academic.oup.com/humrep/article/28/8/2032/658933
- Biason-Lauber A, Konrad D. WNT4 and sex development. Sex Dev. 2008; 2: 210-218. https://www.karger.com/Article/Abstract/152037
- Fontana L, Gentilin B, Fedele L, et al. Genetics of Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome. Clin Genet. 2017; 91: 233-246. https://onlinelibrary.wiley.com/doi/full/10.1111/cge.12883
- Deng S, He Y, Chen N, et al. Spectrum of Type I and Type II Syndromes and Associated Malformations in Chinese Patients with Mayer-Rokitansky-Küster-Hauser Syndrome: A Retrospective Analysis of 274 Cases. J Pediatr Adolesc Gynecol. 2019; 32: 284-287. https://www.jpagonline.org/article/S1083-3188(18)30260-2/fulltext
- Herlin MK, Petersen MB, Brännström M. Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome: a comprehensive update. Orphanet J Rare Dis. 2020; 15: 214. https://ojrd.biomedcentral.com/articles/10.1186/s13023-020-01491-9
- Petrozza JC. Mayer-Rokitansky-Küster-Hauser syndrome and associated malformations: are they as common as we think? Fertil Steril. 2016; 106: 1047-1048. https://www.fertstert.org/article/S0015-0282(16)61388-2/fulltext
- Griffin JE, Edwards C, Madden JD, et al. Congenital absence of the vagina. The Mayer-Rokitansky-Kuster-Hauser syndrome. Ann Intern Med. 1976; 85: 224-236. https://www.acpjournals.org/doi/10.7326/0003-4819-85-2-224
- Opitz JM. Vaginal atresia (von Mayer-Rokitansky-Küster or MRK anomaly) in hereditary renal adysplasia (HRA). Am J Med Genet. 1987; 26: 873-876. https://onlinelibrary.wiley.com/doi/abs/10.1002/ajmg.1320260414
- Guerrier D, Mouchel T, Pasquier L, et al. The Mayer-Rokitansky-Küster-Hauser syndrome (congenital absence of uterus and vagina)--phenotypic manifestations and genetic approaches. J Negat Results Biomed. 2006; 5: 1. https://jnrbm.biomedcentral.com/articles/10.1186/1477-5751-5-1
- Schouten JP, McElgunn CJ, Waaijer R, et al. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002; 30: e57. https://academic.oup.com/nar/article/30/12/e57/2380384
- Bernardini L, Gimelli S, Gervasini C, et al. Recurrent microdeletion at 17q12 as a cause of Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome: two case reports. Orphanet J Rare Dis. 2009; 4: 25. https://ojrd.biomedcentral.com/articles/10.1186/1750-1172-4-25
- Nik-Zainal S, Strick R, Storer M, et al. High incidence of recurrent copy number variants in patients with isolated and syndromic Müllerian aplasia. J Med Genet. 2011; 48: 197-204. https://jmg.bmj.com/content/48/3/197
- Ledig S, Schippert C, Strick R, et al. Recurrent aberrations identified by array-CGH in patients with Mayer-Rokitansky-Küster-Hauser syndrome. Fertil Steril. 2011; 95: 1589-1594. https://www.fertstert.org/article/S0015-0282(10)02168-0/fulltext
- Xia M, Zhao H, Qin Y, et al. LHX1 mutation screening in 96 patients with müllerian duct abnormalities. Fertil Steril. 2012; 97: 682-685. https://www.fertstert.org/article/S0015-0282(11)02857-3/fulltext
- Ledig S, Brucker S, Barresi G, et al. Frame shift mutation of LHX1 is associated with Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome. Hum Reprod. 2012; 27: 2872-2875. https://academic.oup.com/humrep/article/27/9/2872/625703
- Sandbacka M, Laivuori H, Freitas E, et al. TBX6, LHX1 and copy number variations in the complex genetics of Müllerian aplasia. Orphanet J Rare Dis. 2013; 8: 125. https://ojrd.biomedcentral.com/articles/10.1186/1750-1172-8-125
- Williams LS, Demir Eksi D, Shen Y, et al. Genetic analysis of Mayer-Rokitansky-Kuster-Hauser syndrome in a large cohort of families. Fertil Steril. 2017; 108: 145-151. https://www.fertstert.org/article/S0015-0282(17)30398-9/fulltext
- Tewes A-C, Rall KK, Römer T, et al. Variations in RBM8A and TBX6 are associated with disorders of the müllerian ducts. Fertil Steril. 2015; 103: 1313-1318. https://www.fertstert.org/article/S0015-0282(15)00136-3/fulltext
- Pan HX, Luo GN, Wan SQ, et al. Detection of de novo genetic variants in Mayer-Rokitansky-Küster-Hauser syndrome by whole genome sequencing. Eur J Obstet Gynecol Reprod Biol X. 2019; 4: 100089. https://www.sciencedirect.com/science/article/pii/S259016131930122X?via%3Dihub
- Herlin MK, Le VQ, Allan T Højland AT, Ernst A, Okkels H, et al. Whole-exome sequencing identifies a GREB1L variant in a three-generation family with Müllerian and renal agenesis: a novel candidate gene in Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome. A case report. Hum Reprod. 2019; 34: 1838-1846. PubMed: https://pubmed.ncbi.nlm.nih.gov/31424080/
- Jacquinet A, Boujemla B, Fasquelle C, et al. GREB1L variants in familial and sporadic hereditary urogenital adysplasia and Mayer-Rokitansky-Kuster-Hauser syndrome. Clin Genet. 2020; 98: 126-137. https://onlinelibrary.wiley.com/doi/abs/10.1111/cge.13769
- Brophy PD, Rasmussen M, Parida M, et al. A Gene Implicated in Activation of Retinoic Acid Receptor Targets Is a Novel Renal Agenesis Gene in Humans. Genetics. 2017; 207: 215-228. https://academic.oup.com/genetics/article/207/1/215/5930718
- Nakajima T, Sato T, Iguchi T, et al. Retinoic acid signaling determines the fate of the uterus from the mouse Müllerian duct. Reprod Toxicol. 2019; 86: 56-61. https://www.sciencedirect.com/science/article/pii/S0890623818306105?via%3Dihub
- Gervasini C, Grati R, Lalatta F, et al. SHOX duplications found in some cases with type I Mayer-Rokitansky-Kuster-Hauser syndrome. Genet Med. 2010; 12: 634-640. https://www.nature.com/articles/gim2010106
- Sandbacka M, Halttunen M, Jokimaa V, et al. Evaluation of SHOX copy number variations in patients with Müllerian aplasia. Orphanet J Rare Dis. 2011; 6: 53. https://ojrd.biomedcentral.com/articles/10.1186/1750-1172-6-53
- Binder G. Short stature due to SHOX deficiency: genotype, phenotype, and therapy. Horm Res Paediatr. 2011; 75: 81-89. https://www.karger.com/Article/FullText/324105
- Bunyan DJ, Baker KR, Harvey JF, et al. Diagnostic screening identifies a wide range of mutations involving the SHOX gene, including a common 47.5 kb deletion 160 kb downstream with a variable phenotypic effect. Am J Med Genet A. 2013; 161A: 1329-1338. https://onlinelibrary.wiley.com/doi/abs/10.1002/ajmg.a.35919
- Marchini A, Ogata T, Rappold GA. A Track Record on SHOX: From Basic Research to Complex Models and Therapy. Endocr Rev. 2016; 37: 417-448. https://academic.oup.com/edrv/article/37/4/417/2567101
- Dehainault C, Laugé A, Caux-Moncoutier V, et al. Multiplex PCR/liquid chromatography assay for detection of gene rearrangements: application to RB1 gene. Nucleic Acids Res. 2004; 32: e139. https://academic.oup.com/nar/article/32/18/e139/998831
- Bendavid C, Pasquier L, Watrin T, et al. Phenotypic variability of a 4q34-->qter inherited deletion: MRKH syndrome in the daughter, cardiac defect and Fallopian tube cancer in the mother. Eur J Med Genet. 2007; 50: 66-72. https://www.sciencedirect.com/science/article/pii/S1769721206000991?via%3Dihub
- Morcel K, Watrin T, Pasquier L, et al. Utero-vaginal aplasia (Mayer-Rokitansky-Küster-Hauser syndrome) associated with deletions in known DiGeorge or DiGeorge-like loci. Orphanet J Rare Dis. 2011; 6: 9. https://ojrd.biomedcentral.com/articles/10.1186/1750-1172-6-9
- Pontecorvi P, Bernardini L, Capalbo A, et al. Protein-protein interaction network analysis applied to DNA copy number profiling suggests new perspectives on the aetiology of Mayer-Rokitansky-Küster-Hauser syndrome. Sci Rep. 2021; 11: 448. https://www.nature.com/articles/s41598-020-79827-5
- Chen J, Wildhardt G, Zhong Z, et al. Enhancer deletions of the SHOX gene as a frequent cause of short stature: the essential role of a 250 kb downstream regulatory domain. J Med Genet. 2009; 46: 834-839. https://jmg.bmj.com/content/46/12/834
- Benito-Sanz S, Barroso E, Heine-Suner D, et al. Clinical and molecular evaluation of SHOX/PAR1 duplications in Leri-Weill dyschondrosteosis (LWD) and idiopathic short stature (ISS). J Clin Endocrinol Metab. 2011; 96: E404-412. https://doi.org/10.1210/jc.2010-1689
- Rao E, Weiss B, Fukami M, et al. FISH-deletion mapping defines a 270-kb short stature critical interval in the pseudoautosomal region PAR1 on human sex chromosomes. Hum Genet. 1997; 100: 236-239. https://link.springer.com/article/10.1007/s004390050497
- Ogata T, Matsuo N, Nishimura G. SHOX haploinsufficiency and overdosage: impact of gonadal function status. J Med Genet. 2001; 38: 1-6. https://jmg.bmj.com/content/38/1/1
- Benito-Sanz S, Thomas NS, Huber C, et al. A novel class of Pseudoautosomal region 1 deletions downstream of SHOX is associated with Leri-Weill dyschondrosteosis. Am J Hum Genet. 2005; 77: 533-544. https://www.cell.com/ajhg/fulltext/S0002-9297(07)61002-7
- Benito-Sanz S, Belinchon-Martínez A, et al. Identification of 15 novel partial SHOX deletions and 13 partial duplications, and a review of the literature reveals intron 3 to be a hotspot region. J Hum Genet. 2017; 62: 229-234. https://www.nature.com/articles/jhg2016113
- Tiecke E, Bangs F, Blaschke R, et al. Expression of the short stature homeobox gene SHOX is restricted by proximal and distal signals in chick limb buds and affects the length of skeletal elements. Dev Biol. 2006; 298: 585-596. https://www.sciencedirect.com/science/article/pii/S0012160606009900?via%3Dihub
- Thomas NS, Harvey JF, Bunyan DJ, et al. Clinical and molecular characterization of duplications encompassing the human SHOX gene reveal a variable effect on stature. Am J Med Genet A. 2009; 149A: 1407-1414. https://onlinelibrary.wiley.com/doi/abs/10.1002/ajmg.a.32914
- Upners EN, Jensen RB, Rajpert-De Meyts E, et al. Short stature homeobox-containing gene duplications in 3.7% of girls with tall stature and normal karyotypes. Acta Paediatr. 2017; 106: 1651-1657. https://onlinelibrary.wiley.com/doi/abs/10.1111/apa.13969
- Ramirez JM, Rodríguez FA, Echeverría MI, et al. SHOX Duplication and Tall Stature in a Patient with Xq Deletion and Vascular Disease. Case Rep Genet. 2019; 2019: 2691820. https://www.hindawi.com/journals/crig/2019/2691820/
- Fukami M, Naiki Y, Muroya K, et al. Rare pseudoautosomal copy-number variations involving SHOX and/or its flanking regions in individuals with and without short stature. J Hum Genet. 2015; 60: 553-556. https://www.nature.com/articles/jhg201553
- Hirschfeldova K, Solc R. Comparison of SHOX and associated elements duplications distribution between patients (Lėri-Weill dyschondrosteosis/idiopathic short stature) and population sample. Gene. 2017; 627: 164-168. https://www.sciencedirect.com/science/article/pii/S0378111917304845?via%3Dihub
- Tropeano M, Howley D, Gazzellone MJ, et al. Microduplications at the pseudoautosomal SHOX locus in autism spectrum disorders and related neurodevelopmental conditions. J Med Genet. 2016; 53: 536-547. https://jmg.bmj.com/content/53/8/536
- Rall K, Eisenbeis S, Henninger V, et al. Typical and Atypical Associated Findings in a Group of 346 Patients with Mayer-Rokitansky-Kuester-Hauser Syndrome. J Pediatr Adolesc Gynecol. 2015; 28: 362-368. https://www.jpagonline.org/article/S1083-3188(14)00369-6/fulltext
- Kapczuk K, Iwaniec K, Friebe Z, et al. Congenital malformations and other comorbidities in 125 women with Mayer-Rokitansky-Küster-Hauser syndrome. Eur J Obstet Gynecol Reprod Biol. 2016; 207: 45-49. https://www.ejog.org/article/S0301-2115(16)30962-9/fulltext
- Rappold G, Blum WF, Shavrikova EP, et al. Genotypes and phenotypes in children with short stature: clinical indicators of SHOX haploinsufficiency. J Med Genet. 2007; 44: 306-313. https://jmg.bmj.com/content/44/5/306
- Turner DJ, Miretti M, Rajan D, et al. Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nat Genet. 2008; 40: 90-95. https://www.nature.com/articles/ng.2007.40
- May CA, Shone AC, Kalaydjieva L, et al. Crossover clustering and rapid decay of linkage disequilibrium in the Xp/Yp pseudoautosomal gene SHOX. Nat Genet. 2002; 31: 272-275. https://www.nature.com/articles/ng918z
- Lien S, Szyda J, Schechinger B, et al. Evidence for heterogeneity in recombination in the human pseudoautosomal region: high resolution analysis by sperm typing and radiation-hybrid mapping. Am J Hum Genet. 2000; 66: 557-566. https://www.cell.com/ajhg/fulltext/S0002-9297(07)63430-2
- Hastings PJ, Lupski JR, Rosenberg SM, et al. Mechanisms of change in gene copy number. Nat Rev Genet. 2009; 10: 551-564. https://www.nature.com/articles/nrg2593
- Schneider KU, Sabherwal N, Jantz K, et al. Identification of a major recombination hotspot in patients with short stature and SHOX deficiency. Am J Hum Genet. 2005; 77: 89-96. https://www.cell.com/ajhg/fulltext/S0002-9297(07)60904-5
- Newman S, Hermetz KE, Weckselblatt B, et al. Next-generation sequencing of duplication CNVs reveals that most are tandem and some create fusion genes at breakpoints. Am J Hum Genet. 2015; 96: 208-220. https://www.cell.com/ajhg/fulltext/S0002-9297(14)00523-0