
More Information
Submitted: December 05, 2023 | Approved: December 26, 2023 | Published: December 27, 2023
How to cite this article: Somorai MA, Arlt A, Krawitz P, Baumkötter J, Mall V. Individual Treatment Trial of PIGV-Associated Mabry Syndrome with D-Mannose in a Young Child. J Genet Med Gene Ther. 2023; 6: 001-004.
DOI: 10.29328/journal.jgmgt.1001008
Copyright License: © 2023 Somorai MA, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: PIGV-CDG; D-mannose; GPI-anchor biosynthesis deficiencies; PIGV; HPMRS1; Mabry syndrome; Treatment

Individual Treatment Trial of PIGV-Associated Mabry Syndrome with D-Mannose in a Young Child
Marta Agnes Somorai1*, Annabelle Arlt2, Peter Krawitz2, Jochen Baumkötter1 and Volker Mall1
1kbo-Kinderzentrum München, Heiglhofstr. 65, 81377 Munich, Germany
2Institute for Genomic Statistics and Bioinformatics, University of Bonn, 53127 Bonn, Germany
*Address for Correspondence: Marta Agnes Somorai, kbo-Kinderzentrum München, Heiglhofstr. 65, 81377 Munich, Germany, Email: [email protected]
We describe the first individual treatment trial with D-mannose in a young girl with PIGV-CDG. PIGV-CDG belongs to the GPI anchor deficiencies leading to intellectual disability, dysmorphic features, epilepsy, and, less frequently, organ malformations. A hallmark of the GPI anchor deficiencies is the elevated serum alkaline phosphatase (AP). Our patient carried the germline homozygous PIGV variant c.1022C>A, p. (Ala341Glu), the most commonly reported pathogenic variant leading to PIGV-CDG so far. We aimed to improve the impaired enzymatic function of PIGV through elevated substrate levels by giving D-mannose orally. We monitored the clinical status, developmental progress as well as serum AP levels. Our patient experienced no side effects. Standardized developmental testing showed better developmental progress during the 21-months treatment period with D-mannose than in the 12 months following the discontinuation of treatment. The D-Mannose treatment might have had a positive effect on the development of our patient with PIGV-CDG.
Pathogenic biallelic germline variants in PIGV lead to the PIGV-congenital disorder of glycosylation (PIGV-CDG) also known as Hyperphosphatasia with mental retardation syndrome (HPMRS-1, OMIM #239300). It belongs to the genes associated with the clinical entity of Mabry syndrome. Affected individuals exhibit developmental delays, intellectual disability, dysmorphic features (hypertelorism, flat facial profile, brachytelephalangy), elevated serum alkaline phosphatase (AP) (hyperphosphatasia), epilepsy, and less frequently Hirschsprung disease, organ malformations such as renal or anorectal malformations [1].
PIGV encodes the mannosyltransferase II that is involved in the synthesis of the GPI anchor by catalyzing the attachment of the second mannose to the GPI anchor [2]. Germline variants leading to PIGV-CDG involve transcripts with impaired enzyme function [3] leading to deficient GPI anchor synthesis and therefore reduced binding of the GPI anchor substrates. One of these substrates being AP leads to the characteristic elevated serum AP level. Rodríguez de los Santos, et al. established a mouse model for inherited GPI anchor deficiencies with a patient-specific hypomorphic mutation in PIGV. This mouse model exhibits a motor and cognitive phenotype as well as alterations in sociability, mirroring the human phenotype [4].
Our patient was born mature (40 + 4 weeks of gestation) with a slightly low birth weight (2900 g, 7th percentile). At the age of 6 days, she developed a BRUE (brief resolved unexplained event) with cyanosis as well as marked hypotonia and was diagnosed with a hypoplastic aortic arch and aortic isthmus stenosis. She was delayed in her development in all trajectories (walking at the age of 18 months independently, grasping with the full hand, and using 2 meaningful words at the age of 30 months) and showed pronounced restlessness as well as muscular hypotonia. Later, she developed febrile seizures (3 episodes in the 2nd year of life), EEGs showed a high beta activity. NGS panel analysis showed the homozygous variant c.1022C>A, p.(Ala341Glu) in the PIGV gene, the so far in the literature and in ClinVar most commonly reported pathogenic PIGV variant (ClinVar# 1284). Chromosome analysis as well as array CGH were normal. She was presented first to our ambulatory syndrome clinic at the age of 30 months with the typical facial features of patients with Mabry syndrome, including a flat facial profile, prominent sutura metopica, hypertelorism, broad nasal bridge, wide mouth, interdental spaces, and nail dysplasia without brachytelephalangy [5,6]. Fasting serum alkaline-phosphatase (ALP) was elevated: 1008-1144 U/l, mean 1073 U/L, being the key laboratory indicator of the compromised GPI biosynthesis.
The biosynthetic pathway for GPI is mediated by the sequential addition of glycosyl residues and other components to the phosphatidylinositol (PI). Therefore, inborn GPI deficiencies (IGDs) belong to the large group of congenital disorders of glycosylation (CDGs). For 2 types of CDG syndrome, mannose phosphate isomerase- (MPI) and phosphomannomutase 2 (PMM2) deficiency, patients benefit from dietary supplementation of mannose [7-9]. In MPI deficiency, exogenous mannose can bypass the reduced endogenous Man-6-P production through phosphorylation by hexokinase (Figure 1). In PMM2 deficiency, where Man-6-P is the substrate, increasing mannose concentrations can counteract the reduced enzyme activity. Man-1-P, the product of PMM2, is further transformed by GDP-mannose pyrophosphorylase (GMPP) into GDP-mannose, which itself is the substrate of dolichol-phosphate mannosyltransferase (DMP). The product of DMP is Dol-P-Man, which is transferred by mannosyltransferases of the GPI biosynthesis pathway to build the common core of the GPI-anchor [10,11]. Similarly, we hypothesized, that an increased substrate (D-mannose) concentration could result in increased levels of Dol-P-Man in patients with a hypomorphic mutation in PIGV as well, although potentially in a lesser effect, because of the wildtypes in PMM2, GMPP, and DMP in the previous biosynthesis steps.
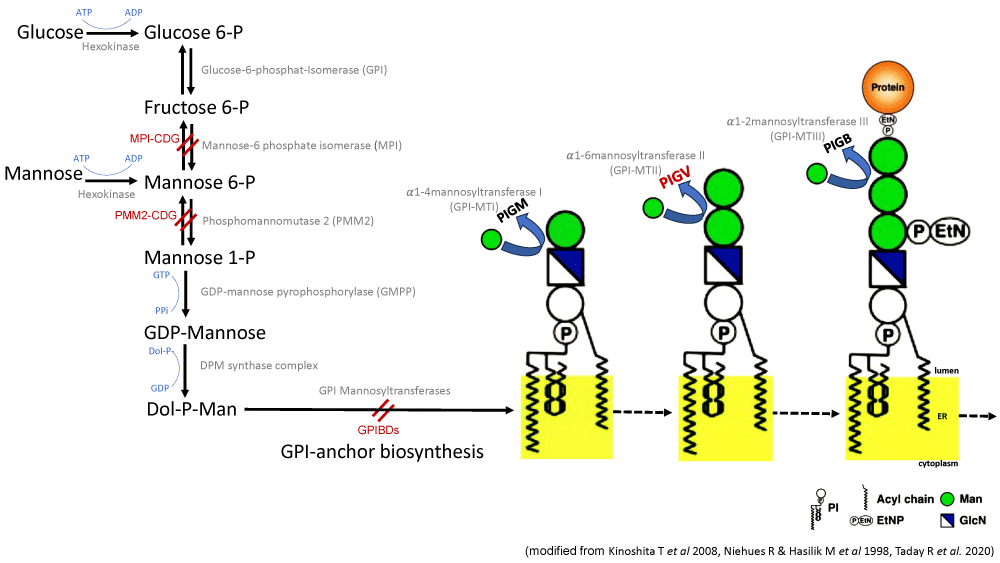
Figure 1: Mannose metabolic pathway and main steps in the GPI anchor biosynthesis featuring mannose residues. For 2 types of CDG syndrome, mannose phosphate isomerase- (MPI) and phosphomannomutase 2 (PMM2) deficiency, mannose can bypass the enzymatic defect and contribute to the GDP-mannose pool. In the further biosynthetic pathway of GPI Dol-P-Man is transferred by the mannosyltransferases I-III (PIGM, PIGV and PIGB) to build the common core of the GPI-anchor.
D-mannose is a widely used substance to prevent cystitis and urinary tract infections. It is available over-the-counter, can be taken orally, and has a generally mild side effect profile with the most common side effect being diarrhea. A serious side effect was published only in association with intravenous administration of high-dose, 1 g/kg/day D-mannose [12]. Also, there is thorough pediatric experience with the usage of D-mannose based on the metabolic treatment of children with MPI and PMM2 [13-15].
After informed consent of the parents we initiated the D-mannose therapy with a slow stepwise dose escalation to avoid diarrhea, starting at 100 mg/kg/day. We targeted 3 doses: 0,6 g/kg/day, 0,8 g/kg/day, and finally 1 g/kg/day. Based on the parental report, our patient was more alert and concentrated in the first few months of treatment receiving 0,6 g/kg/day of D-mannose. She made small developmental steps throughout the 21-months treatment period, followed by a marked slowing of her developmental velocity after discontinuing D-mannose based on the standardized developmental assessments (Münchener Funktionelle Entwicklungsdiagnostik, MFED, Munich functional developmental assessment). At the start of the therapy, she showed a developmental age of 14 - 19 months at the biological age of 30 months. At the end of the D-mannose therapy, she showed a developmental age of 15 - 24 months at the biological age of 50 months. Upon the follow-up 1 year after discontinuation of D-mannose, she showed a developmental age of 15-24 months. She experienced no epileptic seizures throughout the complete period in our care. D-mannose was well tolerated, and no diarrhea or other side effects were observed. The complete treatment period was 21 months. Fasting serum AP was measured repeatedly throughout the treatment trial. The lowest level of AP was 934 U/L (13% decrease from the starting mean level of 1073 U/L), measured under the dose 0,6 g/kg/day after 6 months of administration, and was paradoxically slightly higher under higher doses of D-mannose: 1012 U/L under 0,8 g/kg/day as well as 1034 U/L under 1 g/kg/day (Figure 2).
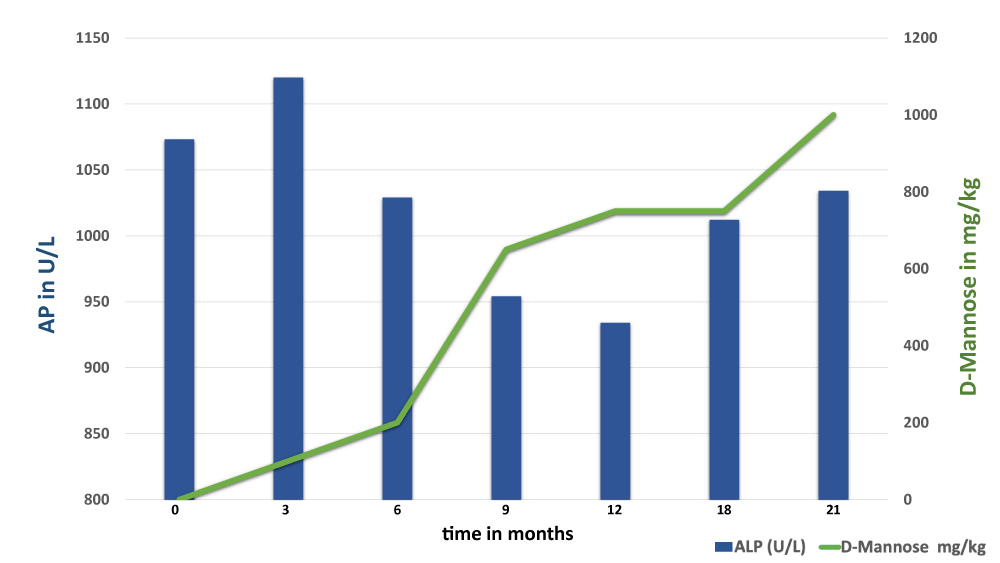
Figure 2: Changes in serum AP in relation to D-Mannose dose given throughout the treatment period. Serum AP showed no clear decrease with higher doses of D-mannose given.
We described the first individual treatment trial with D-mannose in a young patient with PIG-VCDG. There were no side effects reported under treatment. Our patient showed better developmental progress in the 21-months period under D-mannose than in the 12-months period after discontinuing treatment. Long-term D-Mannose treatment might have positive effects on the development of patients with PIGV-CDG. Further studies, especially including more patients as well as regarding dose-finding, are required to allow a clearer judgment on the effect of D-mannose treatment in PIGV-CDG.
- Hutny M, Lipinski P, Jezela-Stanek A. Characteristics of Neuroimaging and Behavioural Phenotype in Polish Patients with PIGV-CDG-An Observational Study and Literature Review. Genes (Basel). 2023 May 31;14(6):1208. doi: 10.3390/genes14061208. PMID: 37372388; PMCID: PMC10298423.
- Kang JY, Hong Y, Ashida H, Shishioh N, Murakami Y, Morita YS, Maeda Y, Kinoshita T. PIG-V involved in transferring the second mannose in glycosylphosphatidylinositol. J Biol Chem. 2005 Mar 11;280(10):9489-97. doi: 10.1074/jbc.M413867200. Epub 2004 Dec 28. PMID: 15623507.
- Horn D, Wieczorek D, Metcalfe K, Barić I, Paležac L, Cuk M, Petković Ramadža D, Krüger U, Demuth S, Heinritz W, Linden T, Koenig J, Robinson PN, Krawitz P. Delineation of PIGV mutation spectrum and associated phenotypes in hyperphosphatasia with mental retardation syndrome. Eur J Hum Genet. 2014 Jun;22(6):762-7. doi: 10.1038/ejhg.2013.241. Epub 2013 Oct 16. PMID: 24129430; PMCID: PMC4023216.
- Rodríguez de Los Santos M, Rivalan M, David FS, Stumpf A, Pitsch J, Tsortouktzidis D, Velasquez LM, Voigt A, Schilling K, Mattei D, Long M, Vogt G, Knaus A, Fischer-Zirnsak B, Wittler L, Timmermann B, Robinson PN, Horn D, Mundlos S, Kornak U, Becker AJ, Schmitz D, Winter Y, Krawitz PM. A CRISPR-Cas9-engineered mouse model for GPI-anchor deficiency mirrors human phenotypes and exhibits hippocampal synaptic dysfunctions. Proc Natl Acad Sci U S A. 2021 Jan 12;118(2):e2014481118. doi: 10.1073/pnas.2014481118. PMID: 33402532; PMCID: PMC7812744.
- Horn D, Krawitz P, Mannhardt A, Korenke GC, Meinecke P. Hyperphosphatasia-mental retardation syndrome due to PIGV mutations: expanded clinical spectrum. Am J Med Genet A. 2011 Aug;155A(8):1917-22. doi: 10.1002/ajmg.a.34102. Epub 2011 Jul 7. PMID: 21739589.
- Krawitz PM, Schweiger MR, Rödelsperger C, Marcelis C, Kölsch U, Meisel C, Stephani F, Kinoshita T, Murakami Y, Bauer S, Isau M, Fischer A, Dahl A, Kerick M, Hecht J, Köhler S, Jäger M, Grünhagen J, de Condor BJ, Doelken S, Brunner HG, Meinecke P, Passarge E, Thompson MD, Cole DE, Horn D, Roscioli T, Mundlos S, Robinson PN. Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat Genet. 2010 Oct;42(10):827-9. doi: 10.1038/ng.653. Epub 2010 Aug 29. PMID: 20802478.
- Čechová A, Altassan R, Borgel D, Bruneel A, Correia J, Girard M, Harroche A, Kiec-Wilk B, Mohnike K, Pascreau T, Pawliński Ł, Radenkovic S, Vuillaumier-Barrot S, Aldamiz-Echevarria L, Couce ML, Martins EG, Quelhas D, Morava E, de Lonlay P, Witters P, Honzík T. Consensus guideline for the diagnosis and management of mannose phosphate isomerase-congenital disorder of glycosylation. J Inherit Metab Dis. 2020 Jul;43(4):671-693. doi: 10.1002/jimd.12241. Epub 2020 Apr 21. PMID: 32266963; PMCID: PMC7574589.
- Niehues R, Hasilik M, Alton G, Körner C, Schiebe-Sukumar M, Koch HG, Zimmer KP, Wu R, Harms E, Reiter K, von Figura K, Freeze HH, Harms HK, Marquardt T. Carbohydrate-deficient glycoprotein syndrome type Ib. Phosphomannose isomerase deficiency and mannose therapy. J Clin Invest. 1998 Apr 1;101(7):1414-20. doi: 10.1172/JCI2350. PMID: 9525984; PMCID: PMC508719.
- Taday R, Grüneberg M, DuChesne I, Reunert J, Marquardt T. Dietary mannose supplementation in phosphomannomutase 2 deficiency (PMM2-CDG). Orphanet J Rare Dis. 2020 Sep 22;15(1):258. doi: 10.1186/s13023-020-01528-z. PMID: 32962735; PMCID: PMC7510076.
- Kinoshita T, Fujita M, Maeda Y. Biosynthesis, remodelling and functions of mammalian GPI-anchored proteins: recent progress. J Biochem. 2008 Sep;144(3):287-94. doi: 10.1093/jb/mvn090. Epub 2008 Jul 17. PMID: 18635593.
- Maeda Y, Murakami Y, Kinoshita T. Synthesis, Genetics, and Congenital Diseases of GPI-Anchored Proteins. In: Kanakura Y, Kinoshita T, Nishimura J-I, editors. Paroxysmal Nocturnal Hemoglobinuria: From Bench to Bedside. Tokyo: Springer Japan; 2017;. 11–54.
- Schroeder AS, Kappler M, Bonfert M, Borggraefe I, Schoen C, Reiter K. Seizures and stupor during intravenous mannose therapy in a patient with CDG syndrome type 1b (MPI-CDG). J Inherit Metab Dis. 2010 Dec;33 Suppl 3:S497-502. doi: 10.1007/s10545-010-9252-x. Epub 2011 Jan 16. PMID: 21240668.
- Rani S, Sahai I, Misra M. A Case of Congenital Disorder of Glycosylation Type 1b Presenting as Hyperinsulinemic Hypoglycemia and Failure to Thrive. JCEM Case Rep. 2023 Sep 21;1(5):luad109. doi: 10.1210/jcemcr/luad109. PMID: 37908211; PMCID: PMC10580457.
- De Graef D, Mousa J, Waberski MB, Morava E. Mannose treatment improves immune deficiency in mannose phosphate isomerase-congenital disorder of glycosylation: case report and review of literature. Ther Adv Rare Dis. 2022 Apr 17;3:26330040221091283. doi: 10.1177/26330040221091283. PMID: 37180423; PMCID: PMC10032425.
- Girard M, Douillard C, Debray D, Lacaille F, Schiff M, Vuillaumier-Barrot S, Dupré T, Fabre M, Damaj L, Kuster A, Torre S, Mention K, McLin V, Dobbelaere D, Borgel D, Bauchard E, Seta N, Bruneel A, De Lonlay P. Long term outcome of MPI-CDG patients on D-mannose therapy. J Inherit Metab Dis. 2020 Nov;43(6):1360-1369. doi: 10.1002/jimd.12289. Epub 2020 Aug 9. PMID: 33098580.