
More Information
Submitted: March 13, 2024 | Approved: March 28, 2024 | Published: March 29, 2024
How to cite this article: Kochumon SP, Nair CKK. Management and Therapeutic Strategies for Spinal Muscular Atrophy. J Genet Med Gene Ther. 2024; 7: 001-007.
DOI: 10.29328/journal.jgmgt.1001009
Copyright License: © 2024 Kochumon SPA, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Spinal Muscular Atrophy (SMA); Supportive care; Survival Motor Neuron (SMN) protein; SMN genes; Carrier analysis-MLPA; Genetic counseling; Prenatal diagnosis; Preimplantation Genetic Diagnosis (PGD); Newborn screening; Disease-Modifying Therapy (DMT); Fetal gene therapy; Gene editing-base editing and CRISPR-Cas

Management and Therapeutic Strategies for Spinal Muscular Atrophy
Sheena P Kochumon1 and Cherupally Krishnan Krishnan Nair2*
1Department of Paediatrics, Amrita Institute of Medical Sciences, Amrita Viswavidyapeeth, Kochi 682041, Kerala, India
2Department of Health Science Research, Amrita Institute of Medical Sciences, Amrita Viswavidyapeeth, Kochi 682041, Kerala, India
*Address for Correspondence: Cherupally Krishnan Krishnan Nair, Department of Health Science Research, Amrita Institute of Medical Sciences, Amrita Viswavidyapeeth, Kochi 682041, Kerala, India, Email: [email protected]; [email protected]
Spinal muscular atrophy is an autosomal recessive neuromuscular disorder characterized by progressive muscle weakness and atrophy. It is one of the most common single-gene disorders with an incidence rate of approximately 1 in 10,000 live births. The clinical manifestations are progressive hypotonia and muscle weakness due to the degeneration of alpha neurons in the anterior horn cells of the spinal cord and motor nuclei in the lower brain stem. Depending on the severity of the symptoms, SMA has five subtypes. Supportive measures can be offered for respiratory, gastrointestinal, and musculoskeletal complications. Carrier testing for all couples is recommended and this can be done by Multiplex Ligation-dependent Probe Amplification (MLPA). Prenatal diagnosis can be offered to carrier couples. Therapies must be given within the newborn period for maximum benefit and before the loss of motor neurons. It is achieved by identifying the SMA babies through Newborn screening. Several new FDA-approved drugs can reduce the progression of symptoms in SMA. However, they cannot offer a definite cure. Clinical follow-up and Neurological assessment demonstrate that SMA children can attain developmental milestones after receiving treatment, which is never normally attained in untreated cases. In utero SMA treatment with Zolgensma would enhance the survival rate and favorable neurological outcomes in the future. Base editing and Gene editing with CRISPR-Cas technologies to target the mutations and restore functional and stable SMN protein levels are the future hopes for a permanent cure of SMA..
SMA is an autosomal recessive neuromuscular disorder characterized by progressive muscle weakness and atrophy and it affects both males and females. SMA was the commonest single gene disorder leading to infant mortality before the FDA approval of disease-modifying treatments [1]. This genetic disorder has an incidence rate of approximately 1 in 6000 - 10,000 live births and with a carrier frequency of 1 in 40 - 60 [1-3]. In the USA, the incidence rate of SMA has been reported to be 1 in 10,000 with a carrier frequency of 1 in 50. A recent Indian study revealed that SMA carrier frequency was 1 in 38 and an incidence of 1 in 10,000 live births [4].
The first two cases of SMA were reported by Austrian neurologist Guido Werdnig in 1891 [5]. It was affirmed as SMA Type 2 after correlating with the current classification of SMA. Several other phenotypes have been reported later. The severest one is fetal-onset form and the mildest is adult-onset form [6,7]. In 1990, the SMA loci were mapped to chromosome 5q13. The genes, SMN1 and SMN2 (SMA-modifier) were identified in 1995. It has led to a great leap in SMA research. Since then diagnosis has been confirmed based on the genetic testing of SMN1 deletion/duplication or mutation instead of muscle biopsy. SMA has been recognized as a progressive neurological disease with high infant mortality since the 19th century; data remains the same even after the introduction of new drugs at the beginning of the 21st century. Several new FDA-approved drugs can reduce the progression of symptoms in SMA. However, they cannot offer a definite cure. In this review, we focus on future management strategies like Base editing and Gene editing with CRISPR-Cas technologies to target the mutations and restore functional and stable SMN protein levels for a permanent cure.
Classification
SMA is classified into five subtypes.
1. Type 0 is the most severe one with prenatal onset and severe hypotonia and respiratory failure soon after birth.
2. Type I Werdnig–Hoffmann disease with hypotonia, symmetrical proximal muscle weakness, inability to sit alone, and onset before 6 months of age.
3. Type II is Dubowitz disease with the inability to stand or walk and onset before 18 months of age.
4. Type III isKugelberg–Welander disease with an onset after 18 months of age.
5. Type IV is the mildest form with onset after 30 years of age.
Clinical features and natural progression of SMA are detailed in Table 1 [6-9].
| Table 1: Types of SMA and their clinical manifestations. | |||||||
| Type of SMA | Percent of total SMA % | Onset of symptoms | Clinical features | Severity | SMN2 copies | Developmental milestones | The course of disease progression |
| 0 | < 1 | Prenatal/ at birth | Generalized weakness, severe hypotonia and respiratory failure, poor feeding, contractures | Most severe | 1 | Not applicable due to early mortality | Death in early infancy |
| 1 | 45 | 0 months - 6 months | Hypotonia, symmetrical proximal muscle weakness, poor feeding, tongue fasciculations, respiratory insufficiency | Severe | 1 - 2 | Nonsitter | Death by 2 years |
| 2 | 20 | 6 months - 18 months | Symmetrical proximal muscle weakness, tongue fasciculations, mini poly myoclonus, scoliosis | Less severe | 3 | Inability to stand Can sit without support | The majority survive up to 25 years |
| 3 | 30 | After 18 months - 30 years | Abnormal gait, proximal lower extremity weakness | Mild | 3 - 4 | Can walk independently | Normal life span |
| 4 | > 5 | > 30 years | Some muscle weakness, tremors and breathing problems | Mildest | 4 or more | Can walk independently | Normal life span |
Multidisciplinary management in SMA
Progressive hypotonia and muscle weakness are the first signs of SMA. It is due to the degeneration of alpha neurons in the anterior horn cells of the Spinal cord and motor nuclei in the lower brain stem. Severe gross motor developmental delay has been noticed in SMA infants. They are prone to develop Pulmonary, Gastrointestinal, and Musculoskeletal complications and psychosocial problems. The quality of life of SMA patients has been achieved through a multidisciplinary approach to management.
Pulmonarycare: Children with SMA have difficulty breathing and coughing due to weak respiratory muscles and rib cage deformities. They are more prone to having sucking and swallowing difficulties and respiratory complications as of bulbar involvement. Facial and ocular muscles are usually spared. Respiratory muscle weakness leads to difficulty in clearing lower respiratory secretions and hypoventilation while sleeping. They are at increased risk of acute respiratory decompensations. The major cause of mortality in infants and children with type 1 and 2 SMA is usually respiratory failure. It’s also mandatory to administer recommended vaccinations to reduce their risk of infection. Chest physiotherapy with postural drainage and early intervention with noninvasive respiratory support is beneficial for SMA type 1 [10-12].
Nutritional care: Aspiration of food and failure to thrive occur because of bulbar dysfunction. It can be manifest as tongue weakness, difficulty in opening the mouth, and poor head control. Other associated problems like gastrointestinal reflux, delayed gastric emptying, and constipation are also seen. Symptoms worsened over a period leading to severe malnutrition.
We can advise changing the consistency of food to increase the intake and thus prevent aspiration. Naso gastric feeding techniques have been advised. Early gastrostomy is another option.
Musculoskeletal care: In immobilized cases, contractures are common. Physiotherapy and spinal bracing can be advised for musculoskeletal complications. It can delay the development of progressive scoliosis that is caused by muscle weakness [13]. The main goal of therapy is to prevent contractures. Other activities such as swimming, aquatic therapy, and adaptive sports are also beneficial.
Manual or motorized wheelchairs can be given as early as 18 months - 24 months of age.
Psychosocial support: Cure SMA India is a parent-led organization for the welfare of SMA families. They are conducting awareness programs to provide psychosocial support and care initiatives on compassionate grounds. They focus on multidisciplinary management through unconditional support to achieve the best possible quality of life and to increase the survival rate.
Genetics of SMA
SMA inheritance: As discussed earlier, SMA is an autosomal recessive neuromuscular disorder. An SMA carrier is an asymptomatic person with a functional copy of the SMN1 gene and an anon-functional copy of SMN1 - ie. (1 + 0) classical carrier. If the partner’s test is negative, the chance of having an affected child is extremely low. If the partner is also a carrier for SMA, there is a 25% risk of having a baby with SMA in each pregnancy. If two parents with no family history of SMA are found to be carriers of routine screening, it is not possible to predict whether they are at risk of having children with the severe form of SMA or one of the less severe forms of SMA. Accordingly, parents of SMA cases are not always carriers, and a small proportion of them are considered de novo or germinal mosaic cases. In that scenario explain the final risk for a given couple. Usually, SMA carrier testing implies the performance of a quantitative method that detects one copy of SMN1 in classical 1/0 carriers. A small proportion of carriers 3% - 4% have two SMN1 copies in cis and none in the other allele 2/0 carriers [14-18].
Chromosomal location (Figure 1): SMA-related genes, SMN1 and SMN2are paralogs and located in q13 region of human chromosome 5. SMN1 gene is located in the telomeric side and SMN2 gene is located in the centromeric side. The SMN1 gene, located in the telomeric side, is the gene responsible for SMA; its loss or defect causes SMA with different phenotypes. The SMN2 gene, located in the centromeric side, is a modifier gene for SMA; its copy number is associated with the severity of the disease.
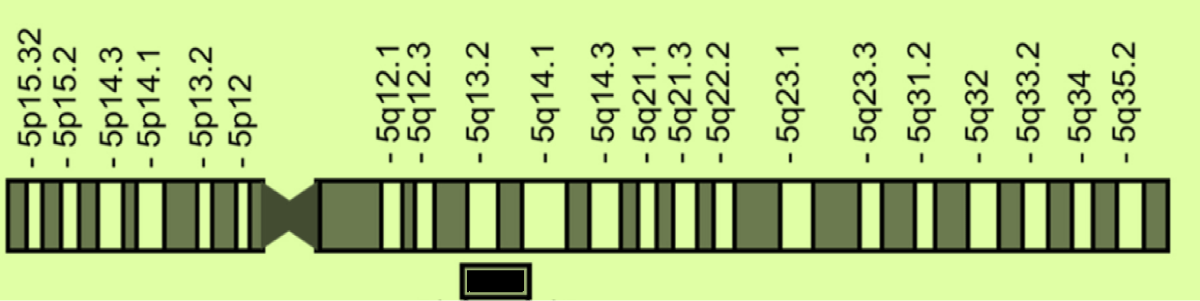
Figure 1: SMN genes in chromosome 5q13. {adapted from [12] Nishio, et al. Int. J. Mol. Sci. 2023, 24, 11939. https://doi.org/ 10.3390/ijms241511939}.
SMA has been confirmed by genetic testing either due to deletion/duplication of the SMN 1 gene or SMN mutation. 95% of cases of SMA are due to a homozygous deletion or mutation of the Survival Motor Neuron 1 (SMN1) gene. SMN1 and the SMA-modifier gene, SMN2 were identified in 1995 [19]. SMN1 gene and SMN2 gene are located within an inverted duplication on chromosome 5q13.2. SMN1 lies telomeric of SMN2.
The SMN1 and SMN2 genes are almost identical and the only difference is a single nucleotide change at the beginning of exon 7 C for SMN1 and T for SMN2 gene. The exon 7 with C nucleotide plays a crucial role in the production of fully functional and stable SMN protein. The exon 7 with T nucleotide in SMN2 cannot fully produce functional and stable SMN protein. Thus SMN1 gene produces 85% to 90% of functional SMN protein and the rest around 10% to 15% by the SMN2 gene [20,21].
The deletions or mutations in the SMN1 gene on chromosome 5q are the most common form of SMA, but rare non-5q SMA cases have also been reported. These point mutations also cause the production of non-functional or unstable SMN proteins. The severity of SMA depends on the number of SMN2 gene copies. More copies of the SMN2 gene mean less severe form of SMA. Four or more copies of SMN2 are present in milder phenotype [22].
While the most common forms of SMA about 95% are caused by deletions or mutations in the SMN1 on chromosome 5q (ie, 5q SMAs), about 5% are compound heterozygotes: they have a deletion or mutation on one of their chromosome 5, and a point mutation on the other chromosome 5, Some rare non-5q spinal muscular atrophies have also been reported [23-25]. The non-5q SMAs are genetically and clinically different.
Genetic testing for spinal muscular atrophy
Carrier testing can be done through a simple blood test. Multiplex ligation-dependent probe amplification (MLPA) is one of the most popular methods used as an initial test as it is convenient, highly sensitive, and capable of determining both SMN1 and SMN2 copy numbers and it aids in the identification of types of SMA. With 95% of affected individuals having a homozygous deletion of SMN1 exon 7, screening for the loss of exon 7 is the first tier in diagnostic testing [26-37]. Another 5% of cases will be caused by other mutations in the SMN1 gene [38,39]. They are compound heterozygotes and have been shown to have a variety of different types of SMN1 mutations including missense mutations [40], nonsense mutations [41], splice site mutations [42,43], insertions, and small deletions. There are rare SMA-affected patients with a single copy of SMN1 and an unidentified second mutation [44-46].
Carrier analysis in family
For asymptomatic siblings of SMA children, genetic testing is offered for age groups more than 18 years. Therefore, genetic counseling is given when they reach reproductive age to ensure that the decision to undergo a genetic test is taken by themselves with adequate information and understanding [47]. If the sibling has one mutated SMN1 copy, then carrier testing must be offered to the partner also. Once the partner also confirms as a carrier, then explain the risk of 25% to their offspring.
Genetic reports are confidential and the genetic counselor/Geneticist can guide the disclosure of the genetic report after getting the patient’s consent.
Prenatal testing for carriers
The American College of Medical Genetics recommends carrier testing for all couples regardless of race or ethnicity [48,49]. The American College of Obstetricians and Gynaecologists (ACOG) emphasized that screening for SMA should be done for all women before pregnancy [50-52].
Detailed genetic counseling is given by a Geneticist/Genetic counselor. During counseling sessions, various options have been given to the couples to make a decision pre-conceptionally and plan future pregnancies.
Genetic counseling: Genetic counseling is recommended for
1. Those who are married in relations – Consanguineous marriage.
2. Those who have a positive family history with SMA or a family member known to be a carrier.
3. An individual without a family history of SMA can be a carrier for this condition as well. About 1 in 40, regardless of ethnic background are also carriers of SMA.
Prenatal diagnosis: It is possible by performing Chorionic villus sampling (CVS) at 11 to 13 weeks or Amniocentesis at 16 to 20 weeks to determine if the fetus is affected or not.
Other options which can be offered are:
1. In Intrauterine Insemination (IUI), sperm from a donor who is not a carrier would be used.
2. In vitro Fertilization (IVF) with their eggs or sperm/ can use donor eggs or sperm.
3. Adoption.
4. PGD-Preimplantation Genetic Diagnosis.
Preimplantation Genetic Diagnosis (PGD) is an option to prevent the transfer of an affected embryo during the IVF.PGD is done on one or two single blastomeres taken from the Day 3 embryo after fertilization [53]. We can select and transfer an unaffected embryo to the uterus.PGD of SMA can be offered to those having an affected child with homozygous exon 7 deletion [54].
Therapeutic approaches
SMA has been considered an incurable disease even at the beginning of the 21st century. Even though the new drugs have reduced the progression of symptoms in SMA, they cannot offer a definite cure.
Recent therapeutic strategies: Disease-modifying therapy (DMT) with nusinersen, onasemnogeneabeparvovec, and risdiplam has been available recently. The Route of administration is different for each therapy.
Nusinersen(Spinraza) is administered by intrathecal injection with maintenance doses once in four months after the loading doses, Loading doses are given as 4 doses, each one in 2 weeks over 8 weeks duration. It is an antisense oligonucleotide that modifies the splicing of the SMN2 gene, thus increasing the availability of functional full-length survival motor neuron protein [55-59].
Risdiplam is administered orally. Risdiplam is a small molecule that alters SMN2 splicing to increase functional SMN protein. It was approved by the FDA in 2020 and by the Drug Controller General of India (DCGI) in 2021. The government of Kerala has started issuing Risdiplam for children less than 5 years of age since July 2022. Clinical follow-up and Neurological assessment demonstrate that SMA children can attain developmental milestones after receiving treatment, which is never normally attained in untreated SMA patients and late-onset patients [60-63].
Gene therapy: Onasemnogeneabeparvovec (Zolgensma) is administered as a single-dose intravenous infusion. Onasemnogeneabeparvovec (Zolgensma) is a gene therapy that utilizes an adeno-associated virus serotype 9 vector to increase the low functional SMN protein levels. In 2019, the U.S. Food and Drug Administration approved Zolgensma, which alters the underlying genetic cause of spinal muscular atrophy and helps to stop the disease’s progression. It has been approved for treating SMA cases with homozygous mutation in SMN1 [64,65].
Future perspectives
Therapies must be given within the newborn period for maximum benefit and before the loss of motor neurons. It can achieved by identifying the SMA in infants through Newborn screening. Newborn Screening (NBS) is intended to find out those newborns who are at increased risk of certain genetic disorders. Newborn screening can be done on dried blood samples to confirm homozygous deletion of exon 7 in SMN1. The most beneficial response to SMA treatments has been documented for the treatment of pre-symptomatic SMA cases. Newborn screening and adequate pre-test genetic counseling are essential for providing precise information to the families. In utero SMA treatment with Zolgensma would enhance the survival rate and favorable neurological outcomes in the future. We can assess the acceptance of parents with SMA for prenatal diagnosis, fetal therapies with gene therapy being favored, and clinical trials. Future research and follow-up are needed to understand the long-term outcome of treatment with single therapy or combination therapies [66-68].
Gene editing - Innovative therapeutic strategy (CRISPR and base editing)
The Clustered Regularly Interspaced Short Palindromic Repeat sequences (CRISPR) and the associated Cas nuclease system are the most popular genome editing platforms due to their simplicity, cost-effectiveness, and specificity in gene editing by introducing specific nucleotides. The mutated DNA sequence is edited with a guide RNA – Cas protein complex and this complex is introduced into the cells for gene editing with a viral vector or a nanoparticle. The guide RNA will have some length of homology with the gene to be edited/corrected. CRISPR/Cas facilitates nonhomologous end joining, homology-directed synthesis, nonhomologous end joining, and single base exchanges. At present a variety of CRISPR/Cas based therapeutics are being used in gene therapy in several human genetic diseases. Pluripotent stem cells of placental origin or extracted from patients following gene editing using CRISPR/Cas can be reintroduced into the patients [69]. Great progress has been made in recent times in genome editing. Apart from CRISPR/Cas, several nucleases capable of genome editing like zinc finger nuclease, transcription-activated effector nucleases, etc. are available. However, they lack the versatility, simplicity, and utility of gene therapy. Before undertaking gene manipulation therapy, several issues such as the ethical, legal, and social implications of gene editing technologies should be considered.
A recent study at Harvard Medical School and Massachusetts General Hospital showed that fibroblasts from SMA patients as well as a mouse model of the disease could have their levels of the Survival Motor Neuron (SMN) protein restored by using Adenosine Base Editors (ABEs). The results proved that using base editing or other CRISPR-based techniques to target the genetic mutations restored functional and stable SMN protein levels leading to permanent cure in SMA patients [70].
- Groen EJN, Talbot K, Gillingwater TH. Advances in therapy for spinal muscular atrophy: promises and challenges. Nat Rev Neurol. 2018 Apr;14(4):214-224. doi: 10.1038/nrneurol.2018.4. Epub 2018 Feb 9. PMID: 29422644.
- Phan HC, Taylor JL, Hannon H, Howell R. Newborn screening for spinal muscular atrophy: Anticipating an imminent need. Semin Perinatol. 2015 Apr;39(3):217-29. doi: 10.1053/j.semperi.2015.03.006. PMID: 25979781.
- Ogino S, Wilson RB. Genetic testing and risk assessment for spinal muscular atrophy (SMA). Hum Genet. 2002 Dec;111(6):477-500. doi: 10.1007/s00439-002-0828-x. Epub 2002 Oct 3. PMID: 12436240.
- Nilay M, Moirangthem A, Saxena D, Mandal K, Phadke SR. Carrier frequency of SMN1-related spinal muscular atrophy in north Indian population: The need for population based screening program. Am J Med Genet A. 2021 Jan;185(1):274-277. doi: 10.1002/ajmg.a.61918. Epub 2020 Oct 14. PMID: 33051992.
- Werdnig G. ZweifrühinfantilehereditäreFälle von progressiverMuskelatrophieunterdemBilde der Dystrophie, aberanfneurotischerGrundlage. Arch. FürPsychiatr. Nervenkrankh. 1891; 22: 437-480.
- Dubowitz V. Very severe spinal muscular atrophy (SMA type 0): an expanding clinical phenotype. Eur J Paediatr Neurol. 1999;3(2):49-51. doi: 10.1053/ejpn.1999.0181. PMID: 10700538.
- Clermont O, Burlet P, Lefebvre S, Bürglen L, Munnich A, Melki J. SMN gene deletions in adult-onset spinal muscular atrophy. Lancet. 1995 Dec 23-30;346(8991-8992):1712-3. doi: 10.1016/s0140-6736(95)92881-2. PMID: 8551862.
- Thomas NH, Dubowitz V. The natural history of type I (severe) spinal muscular atrophy. Neuromuscul Disord. 1994 Sep-Nov;4(5-6):497-502. doi: 10.1016/0960-8966(94)90090-6. PMID: 7881295.
- Arnold WD, Kassar D, Kissel JT. Spinal muscular atrophy: diagnosis and management in a new therapeutic era. Muscle Nerve. 2015 Feb;51(2):157-67. doi: 10.1002/mus.24497. Epub 2014 Dec 16. PMID: 25346245; PMCID: PMC4293319.
- Schroth MK. Special considerations in the respiratory management of spinal muscular atrophy. Pediatrics. 2009 May;123 Suppl 4:S245-9. doi: 10.1542/peds.2008-2952K. PMID: 19420154.
- Bach JR, Niranjan V, Weaver B. Spinal muscular atrophy type 1: A noninvasive respiratory management approach. Chest. 2000 Apr;117(4):1100-5. doi: 10.1378/chest.117.4.1100. PMID: 10767247.
- Nishio H, Niba ETE, Saito T, Okamoto K, Takeshima Y, Awano H. Spinal Muscular Atrophy: The Past, Present, and Future of Diagnosis and Treatment. Int J Mol Sci. 2023 Jul 26;24(15):11939. doi: 10.3390/ijms241511939. PMID: 37569314; PMCID: PMC10418635.
- Tangsrud SE, Carlsen KC, Lund-Petersen I, Carlsen KH. Lung function measurements in young children with spinal muscle atrophy; a cross sectional survey on the effect of position and bracing. Arch Dis Child. 2001 Jun;84(6):521-4. doi: 10.1136/adc.84.6.521. PMID: 11369575; PMCID: PMC1718814.
- Bernal S, Also-Rallo E, Martínez-Hernández R, Alías L, Rodríguez-Alvarez FJ, Millán JM, Hernández-Chico C, Baiget M, Tizzano EF. Plastin 3 expression in discordant spinal muscular atrophy (SMA) siblings. Neuromuscul Disord. 2011 Jun;21(6):413-9. doi: 10.1016/j.nmd.2011.03.009. Epub 2011 May 4. PMID: 21546251.
- Alías L, Barceló MJ, Bernal S, Martínez-Hernández R, Also-Rallo E, Vázquez C, Santana A, Millán JM, Baiget M, Tizzano EF. Improving detection and genetic counseling in carriers of spinal muscular atrophy with two copies of the SMN1 gene. Clin Genet. 2014 May;85(5):470-5. doi: 10.1111/cge.12222. Epub 2013 Jul 16. PMID: 23799925.
- Luo M, Liu L, Peter I, Zhu J, Scott SA, Zhao G, Eversley C, Kornreich R, Desnick RJ, Edelmann L. An Ashkenazi Jewish SMN1 haplotype specific to duplication alleles improves pan-ethnic carrier screening for spinal muscular atrophy. Genet Med. 2014 Feb;16(2):149-56. doi: 10.1038/gim.2013.84. Epub 2013 Jun 20. PMID: 23788250.
- Alías L, Bernal S, Calucho M, Martínez E, March F, Gallano P, Fuentes-Prior P, Abuli A, Serra-Juhe C, Tizzano EF. Utility of two SMN1 variants to improve spinal muscular atrophy carrier diagnosis and genetic counselling. Eur J Hum Genet. 2018 Oct;26(10):1554-1557. doi: 10.1038/s41431-018-0193-4. Epub 2018 Jun 14. PMID: 29904179; PMCID: PMC6138687.
- Jedrzejowska M, Borkowska J, Zimowski J, Kostera-Pruszczyk A, Milewski M, Jurek M, Sielska D, Kostyk E, Nyka W, Zaremba J, Hausmanowa-Petrusewicz I. Unaffected patients with a homozygous absence of the SMN1 gene. Eur J Hum Genet. 2008 Aug;16(8):930-4. doi: 10.1038/ejhg.2008.41. Epub 2008 Mar 12. PMID: 18337729.
- Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995 Jan 13;80(1):155-65. doi: 10.1016/0092-8674(95)90460-3. PMID: 7813012.
- Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci U S A. 1999 May 25;96(11):6307-11. doi: 10.1073/pnas.96.11.6307. PMID: 10339583; PMCID: PMC26877.
- Monani UR, Lorson CL, Parsons DW, Prior TW, Androphy EJ, Burghes AH, McPherson JD. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet. 1999 Jul;8(7):1177-83. doi: 10.1093/hmg/8.7.1177. PMID: 10369862.
- Mailman MD, Heinz JW, Papp AC. Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet Med 2002; 4:20.Darras BT. Non-5q spinal muscular atrophies: the alphanumeric soup thickens. Neurology. 2011; 77:312.
- Darras BT. Non-5q spinal muscular atrophies: the alphanumeric soup thickens. Neurology. 2011 Jul 26;77(4):312-4. doi: 10.1212/WNL.0b013e3182267bd8. Epub 2011 Jun 29. PMID: 21715708.
- Zerres K, Rudnik-Schöneborn S. 93rd ENMC international workshop: non-5q-spinal muscular atrophies (SMA) - clinical picture (6-8 April 2001, Naarden, The Netherlands). Neuromuscul Disord. 2003 Feb;13(2):179-83. doi: 10.1016/s0960-8966(02)00211-0. PMID: 12565918.
- Peeters K, Chamova T, Jordanova A. Clinical and genetic diversity of SMN1-negative proximal spinal muscular atrophies. Brain. 2014 Nov;137(Pt 11):2879-96. doi: 10.1093/brain/awu169. Epub 2014 Jun 25. PMID: 24970098; PMCID: PMC4208460.
- Wirth B, Karakaya M, Kye MJ, Mendoza-Ferreira N. Twenty-Five Years of Spinal Muscular Atrophy Research: From Phenotype to Genotype to Therapy, and What Comes Next. Annu Rev Genomics Hum Genet. 2020 Aug 31;21:231-261. doi: 10.1146/annurev-genom-102319-103602. Epub 2020 Jan 31. PMID: 32004094.
- Dubowitz V. Very severe spinal muscular atrophy (SMA type 0): an expanding clinical phenotype. Eur J Paediatr Neurol. 1999;3(2):49-51. doi: 10.1053/ejpn.1999.0181. PMID: 10700538.
- Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995 Jan 13;80(1):155-65. doi: 10.1016/0092-8674(95)90460-3. PMID: 7813012.
- Bürglen L, Lefebvre S, Clermont O, Burlet P, Viollet L, Cruaud C, Munnich A, Melki J. Structure and organization of the human survival motor neurone (SMN) gene. Genomics. 1996 Mar 15;32(3):479-82. doi: 10.1006/geno.1996.0147. PMID: 8838816.
- McAndrew PE, Parsons DW, Simard LR, Rochette C, Ray PN, Mendell JR, Prior TW, Burghes AH. Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am J Hum Genet. 1997 Jun;60(6):1411-22. doi: 10.1086/515465. PMID: 9199562; PMCID: PMC1716150.
- Mailman MD, Heinz JW, Papp AC, Snyder PJ, Sedra MS, Wirth B, Burghes AH, Prior TW. Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet Med. 2002 Jan-Feb;4(1):20-6. doi: 10.1097/00125817-200201000-00004. PMID: 11839954.
- Crawford TO, Paushkin SV, Kobayashi DT, Forrest SJ, Joyce CL, Finkel RS, Kaufmann P, Swoboda KJ, Tiziano D, Lomastro R, Li RH, Trachtenberg FL, Plasterer T, Chen KS; Pilot Study of Biomarkers for Spinal Muscular Atrophy Trial Group. Evaluation of SMN protein, transcript, and copy number in the biomarkers for spinal muscular atrophy (BforSMA) clinical study. PLoS One. 2012;7(4):e33572. doi: 10.1371/journal.pone.0033572. Epub 2012 Apr 27. PMID: 22558076; PMCID: PMC3338744.
- Wadman RI, Stam M, Gijzen M, Lemmink HH, Snoeck IN, Wijngaarde CA, Braun KP, Schoenmakers MA, van den Berg LH, Dooijes D, van der Pol WL. Association of motor milestones, SMN2 copy and outcome in spinal muscular atrophy types 0-4. J Neurol Neurosurg Psychiatry. 2017 Apr;88(4):365-367. doi: 10.1136/jnnp-2016-314292. Epub 2017 Jan 20. PMID: 28108522.
- Calucho M, Bernal S, Alías L, March F, Venceslá A, Rodríguez-Álvarez FJ, Aller E, Fernández RM, Borrego S, Millán JM, Hernández-Chico C, Cuscó I, Fuentes-Prior P, Tizzano EF. Correlation between SMA type and SMN2 copy number revisited: An analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul Disord. 2018 Mar;28(3):208-215. doi: 10.1016/j.nmd.2018.01.003. Epub 2018 Jan 11. PMID: 29433793.
- Prior TW, Swoboda KJ, Scott HD, Hejmanowski AQ. Homozygous SMN1 deletions in unaffected family members and modification of the phenotype by SMN2. Am J Med Genet A. 2004 Oct 15;130A(3):307-10. doi: 10.1002/ajmg.a.30251. PMID: 15378550; PMCID: PMC4349519.
- Monani UR, Coovert DD, Burghes AH. Animal models of spinal muscular atrophy. Hum Mol Genet. 2000 Oct;9(16):2451-7. doi: 10.1093/hmg/9.16.2451. PMID: 11005801.
- Prior TW, Krainer AR, Hua Y, Swoboda KJ, Snyder PC, Bridgeman SJ, Burghes AH, Kissel JT. A positive modifier of spinal muscular atrophy in the SMN2 gene. Am J Hum Genet. 2009 Sep;85(3):408-13. doi: 10.1016/j.ajhg.2009.08.002. Epub 2009 Aug 27. PMID: 19716110; PMCID: PMC2771537.
- Bernal S, Alías L, Barceló MJ, Also-Rallo E, Martínez-Hernández R, Gámez J, Guillén-Navarro E, Rosell J, Hernando I, Rodríguez-Alvarez FJ, Borrego S, Millán JM, Hernández-Chico C, Baiget M, Fuentes-Prior P, Tizzano EF. The c.859G>C variant in the SMN2 gene is associated with types II and III SMA and originates from a common ancestor. J Med Genet. 2010 Sep;47(9):640-2. doi: 10.1136/jmg.2010.079004. Epub 2010 Jun 24. PMID: 20577007.
- Yamamoto T, Sato H, Lai PS, Nurputra DK, Harahap NI, Morikawa S, Nishimura N, Kurashige T, Ohshita T, Nakajima H, Yamada H, Nishida Y, Toda S, Takanashi J, Takeuchi A, Tohyama Y, Kubo Y, Saito K, Takeshima Y, Matsuo M, Nishio H. Intragenic mutations in SMN1 may contribute more significantly to clinical severity than SMN2 copy numbers in some spinal muscular atrophy (SMA) patients. Brain Dev. 2014 Nov;36(10):914-20. doi: 10.1016/j.braindev.2013.11.009. Epub 2013 Dec 17. PMID: 24359787.
- Mendonça RH, Matsui C Jr, Polido GJ, Silva AMS, Kulikowski L, Torchio Dias A, Zanardo EA, Solla DJF, Gurgel-Giannetti J, de Moura ACML, Sampaio GPC, Oliveira ASB, de Souza PVS, Pinto WBVR, Gonçalves EA, Farias IB, Nardes F, Araújo APQC, Marques W Jr, Tomaselli PJ, Ribeiro MDO, Kitajima JP, Paoli Monteiro F, Saute JAM, Becker MM, Saraiva-Pereira ML, Brusius-Facchin AC, van der Linden V, Florêncio RN, Barbosa AVS, Machado-Costa MC, Pessoa ALS, Souza LS, Franca MC Jr, Kok F, Reed UC, Zanoteli E. Intragenic variants in the SMN1 gene determine the clinical phenotype in 5q spinal muscular atrophy. Neurol Genet. 2020 Sep 1;6(5):e505. doi: 10.1212/NXG.0000000000000505. PMID: 33062891; PMCID: PMC7524579.
- Wu X, Wang SH, Sun J, Krainer AR, Hua Y, Prior TW. A-44G transition in SMN2 intron 6 protects patients with spinal muscular atrophy. Hum Mol Genet. 2017 Jul 15;26(14):2768-2780. doi: 10.1093/hmg/ddx166. PMID: 28460014; PMCID: PMC5886194.
- Hofmann Y, Lorson CL, Stamm S, Androphy EJ, Wirth B. Htra2-beta 1 stimulates an exonic splicing enhancer and can restore full-length SMN expression to survival motor neuron 2 (SMN2). Proc Natl Acad Sci U S A. 2000 Aug 15;97(17):9618-23. doi: 10.1073/pnas.160181697. PMID: 10931943; PMCID: PMC16914.
- Oprea GE, Kröber S, McWhorter ML, Rossoll W, Müller S, Krawczak M, Bassell GJ, Beattie CE, Wirth B. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science. 2008 Apr 25;320(5875):524-7. doi: 10.1126/science.1155085. PMID: 18440926; PMCID: PMC4908855.
- McGovern VL, Massoni-Laporte A, Wang X, Le TT, Le HT, Beattie CE, Rich MM, Burghes AH. Plastin 3 Expression Does Not Modify Spinal Muscular Atrophy Severity in the ∆7 SMA Mouse. PLoS One. 2015 Jul 2;10(7):e0132364. doi: 10.1371/journal.pone.0132364. PMID: 26134627; PMCID: PMC4489873.
- Riessland M, Kaczmarek A, Schneider S, Swoboda KJ, Löhr H, Bradler C, Grysko V, Dimitriadi M, Hosseinibarkooie S, Torres-Benito L, Peters M, Upadhyay A, Biglari N, Kröber S, Hölker I, Garbes L, Gilissen C, Hoischen A, Nürnberg G, Nürnberg P, Walter M, Rigo F, Bennett CF, Kye MJ, Hart AC, Hammerschmidt M, Kloppenburg P, Wirth B. Neurocalcin Delta Suppression Protects against Spinal Muscular Atrophy in Humans and across Species by Restoring Impaired Endocytosis. Am J Hum Genet. 2017 Feb 2;100(2):297-315. doi: 10.1016/j.ajhg.2017.01.005. Epub 2017 Jan 26. PMID: 28132687; PMCID: PMC5294679.
- Bernal S, Also-Rallo E, Martínez-Hernández R, Alías L, Rodríguez-Alvarez FJ, Millán JM, Hernández-Chico C, Baiget M, Tizzano EF. Plastin 3 expression in discordant spinal muscular atrophy (SMA) siblings. Neuromuscul Disord. 2011 Jun;21(6):413-9. doi: 10.1016/j.nmd.2011.03.009. Epub 2011 May 4. PMID: 21546251.
- Borry P, Evers-Kiebooms G, Cornel MC, Clarke A, Dierickx K. Public and Professional Policy Committee [PPPC] of the European Society of Human Genetics [ESHG]. Genetic testing in asymptomatic minors: background considerations towards ESHG Recommendations. Eur J Hum Genet. 2009; 17:711-9.
- Prior TW; Professional Practice and Guidelines Committee. Carrier screening for spinal muscular atrophy. Genet Med. 2008 Nov;10(11):840-2. doi: 10.1097/GIM.0b013e318188d069. PMID: 18941424; PMCID: PMC3110347.
- Prior TW. Spinal muscular atrophy: a time for screening. Curr Opin Pediatr. 2010 Dec;22(6):696-702. doi: 10.1097/MOP.0b013e32833f3046. PMID: 20829691.
- Committee Opinion No. 691: Carrier Screening for Genetic Conditions. Obstet Gynecol. 2017 Mar;129(3):e41-e55. doi: 10.1097/AOG.0000000000001952. PMID: 28225426.
- Prior TW; Professional Practice and Guidelines Committee. Carrier screening for spinal muscular atrophy. Genet Med. 2008 Nov;10(11):840-2. doi: 10.1097/GIM.0b013e318188d069. PMID: 18941424; PMCID: PMC3110347.
- Prior TW, Nagan N, Sugarman EA, Batish SD, Braastad C. Technical standards and guidelines for spinal muscular atrophy testing. Genet Med. 2011 Jul;13(7):686-94. doi: 10.1097/GIM.0b013e318220d523. PMID: 21673580.
- Hardy K, Handyside AH. Biopsy of cleavage stage human embryos and diagnosis of single gene defects by DNA amplification. Arch Pathol Lab Med. 1992 Apr;116(4):388-92. PMID: 1558477.
- Lee I, Alur-Gupta S, Gallop R, Dokras A. Utilization of preimplantation genetic testing for monogenic disorders. Fertil Steril. 2020 Oct;114(4):854-860. doi: 10.1016/j.fertnstert.2020.05.045. PMID: 33040985.
- Ramdas S, Servais L. New treatments in spinal muscular atrophy: an overview of currently available data. Expert Opin Pharmacother. 2020 Feb;21(3):307-315. doi: 10.1080/14656566.2019.1704732. PMID: 31973611.
- Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, Prior TW, Lowes L, Alfano L, Berry K, Church K, Kissel JT, Nagendran S, L'Italien J, Sproule DM, Wells C, Cardenas JA, Heitzer MD, Kaspar A, Corcoran S, Braun L, Likhite S, Miranda C, Meyer K, Foust KD, Burghes AHM, Kaspar BK. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N Engl J Med. 2017 Nov 2;377(18):1713-1722. doi: 10.1056/NEJMoa1706198. PMID: 29091557.
- Finkel RS, Mercuri E, Darras BT, Connolly AM, Kuntz NL, Kirschner J, Chiriboga CA, Saito K, Servais L, Tizzano E, Topaloglu H, Tulinius M, Montes J, Glanzman AM, Bishop K, Zhong ZJ, Gheuens S, Bennett CF, Schneider E, Farwell W, De Vivo DC; ENDEAR Study Group. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N Engl J Med. 2017 Nov 2;377(18):1723-1732. doi: 10.1056/NEJMoa1702752. PMID: 29091570.
- Mercuri E, Darras BT, Chiriboga CA, Day JW, Campbell C, Connolly AM, Iannaccone ST, Kirschner J, Kuntz NL, Saito K, Shieh PB, Tulinius M, Mazzone ES, Montes J, Bishop KM, Yang Q, Foster R, Gheuens S, Bennett CF, Farwell W, Schneider E, De Vivo DC, Finkel RS; CHERISH Study Group. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N Engl J Med. 2018 Feb 15;378(7):625-635. doi: 10.1056/NEJMoa1710504. PMID: 29443664.
- Haché M, Swoboda KJ, Sethna N, Farrow-Gillespie A, Khandji A, Xia S, Bishop KM. Intrathecal Injections in Children With Spinal Muscular Atrophy: Nusinersen Clinical Trial Experience. J Child Neurol. 2016 Jun;31(7):899-906. doi: 10.1177/0883073815627882. Epub 2016 Jan 27. PMID: 26823478; PMCID: PMC4871174.
- Seabrook T, Baranello G, Servais L. FIREFISH part 1: early clinical results following an increase of SMN protein in infants with type 1 spinal muscular atrophy (SMA) treated with risdiplam (RG7916). Paper presented at: MDA Clinical & Scientific Conference. 2019. Orlando.
- Baranello G, Servais L, Day J. FIREFISH part 1: 16-month safety and exploratory outcomes of risdiplam (RG7916) treatment in infants with type 1 spinal muscular atrophy. NeuromusculDisord. 2019; 29:S184; 353. doi:10.1016/j.nmd.2019.06.515.
- Chiriboga CA, Mercuri E, Fischer D. JEWELFISH: safety and pharmacodynamic data in patients with spinal muscular atrophy (SMA) receiving treatment with risdiplam (RG7916) that have previously been treated with nusinersen. NeuromusculDisord. 2019; 29:S187; 363. doi:10.1016/j.nmd.2019.06.525.
- De Vivo DC, Bertini E, Swoboda KJ, Hwu WL, Crawford TO, Finkel RS, Kirschner J, Kuntz NL, Parsons JA, Ryan MM, Butterfield RJ, Topaloglu H, Ben-Omran T, Sansone VA, Jong YJ, Shu F, Staropoli JF, Kerr D, Sandrock AW, Stebbins C, Petrillo M, Braley G, Johnson K, Foster R, Gheuens S, Bhan I, Reyna SP, Fradette S, Farwell W; NURTURE Study Group. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: Interim efficacy and safety results from the Phase 2 NURTURE study. Neuromuscul Disord. 2019 Nov;29(11):842-856. doi: 10.1016/j.nmd.2019.09.007. Epub 2019 Sep 12. PMID: 31704158; PMCID: PMC7127286.
- Ogbonmide T, Rathore R, Rangrej SB, Hutchinson S, Lewis M, Ojilere S, Carvalho V, Kelly I. Gene Therapy for Spinal Muscular Atrophy (SMA): A Review of Current Challenges and Safety Considerations for Onasemnogene Abeparvovec (Zolgensma). Cureus. 2023 Mar 15;15(3):e36197. doi: 10.7759/cureus.36197. PMID: 37065340; PMCID: PMC10104684.
- Atsumi A, Yoneda T, Tsuchida K, Kagawa Y, Tominaga S, Kawase K, Kikuchi N. [Pharmacological and clinical profile of Onasemnogene Aveparvovec, the first gene therapy for spinal muscular atrophy (SMA)]. Nihon Yakurigaku Zasshi. 2022;157(1):53-61. Japanese. doi: 10.1254/fpj.21066. PMID: 34980814.
- Chiriboga CA, Mercuri E, Fischer D, et al. P.363JEWELFISH: safety and pharmacodynamic data in patients with spinal muscular atrophy (SMA) receiving treatment with risdiplam (RG7916) that have previously been treated with nusinersen. NeuromusculDisord. 2019; 29:S187. doi:10.1016/j.nmd.2019.06.525.
- Bertini E, Day J, Muhaizea M, et al. P.362RAINBOWFISH: a study of risdiplam (RG7916) in newborns with pre-symptomatic spinal muscular atrophy (SMA). NeuromusculDisord. 2019; 29:S187. doi:10.1016/j.nmd.2019.06.524.
- Chen TH. New and Developing Therapies in Spinal Muscular Atrophy: From Genotype to Phenotype to Treatment and Where Do We Stand? Int J Mol Sci. 2020 May 7;21(9):3297. doi: 10.3390/ijms21093297. PMID: 32392694; PMCID: PMC7246502.
- Corti S, Nizzardo M, Nardini M, Donadoni C, Salani S, Ronchi D, Simone C, Falcone M, Papadimitriou D, Locatelli F, Mezzina N, Gianni F, Bresolin N, Comi GP. Embryonic stem cell-derived neural stem cells improve spinal muscular atrophy phenotype in mice. Brain. 2010 Feb;133(Pt 2):465-81. doi: 10.1093/brain/awp318. Epub 2009 Dec 23. PMID: 20032086.
- Alves CRR, Ha LL, Yaworski R, Sutton ER, Lazzarotto CR, Christie KA, Reilly A, Beauvais A, Doll RM, de la Cruz D, Maguire CA, Swoboda KJ, Tsai SQ, Kothary R, Kleinstiver BP. Optimization of base editors for the functional correction of SMN2 as a treatment for spinal muscular atrophy. Nat Biomed Eng. 2024 Feb;8(2):118-131. doi: 10.1038/s41551-023-01132-z. Epub 2023 Dec 6. PMID: 38057426; PMCID: PMC10922509.