
More Information
Submitted: April 23, 2025 | Approved: April 30, 2025 | Published: May 01, 2025
How to cite this article: Asadi S, Zare A, Koohestani S. The Role of Genetic Mutations in the HPGD & SLCO2A1 Genes in Pachydermoperiostosis Syndrome. J Genet Med Gene Ther. 2025; 8(1): 001-005. Available from:
https://dx.doi.org/10.29328/journal.jgmgt.1001013.
DOI: 10.29328/journal.jgmgt.1001013
Copyright License: © 2025 Asadi S, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Pachydermoperiostosis syndrome; Genetic disorder; Mutated genes; HPGD gene; SLCO2A1 gene

The Role of Genetic Mutations in the HPGD & SLCO2A1 Genes in Pachydermoperiostosis Syndrome
Shahin Asadi* , Arezo Zare and Sima Koohestani
, Arezo Zare and Sima Koohestani
Medical Genetics Director of the Division of Medical Genetics and Molecular Pathology Research, Division of Medical Genetics and Molecular Pathology Research, Center of Complex Disease, USA
*Address for Correspondence: Shahin Asadi, Medical Genetics Director of the Division of Medical Genetics and Molecular Pathology Research, Division of Medical Genetics and Molecular Pathology Research, Center of Complex Disease, USA, Email: [email protected]
Pachydermoperiostosis, also known as Primary Hypertrophic Osteoarthropathy (PHO), is a rare genetic disorder. The three main features are: enlarged fingertips (clubbing), thickened facial skin (pachydermia), and excessive sweating (hyperhidrosis). PHO is characterized by problems with skin and bone growth. Patients with PHO usually have coarse facial features with oily, thick, grooved skin on the face, joint pain, enlarged fingertips and toes, and hyperhidrosis of the hands and feet. Symptoms vary individually; however, men generally present with more severe manifestations. X-rays can help check for features that are not noticeable to the naked eye. There are two genes that are associated with PHO: the HPGD gene, located on the long arm of chromosome 4 at 4q34.1, and the SLCO2A1 gene, located on the long arm of chromosome 3 at 3q22.1 - q22.2. Mutations in the HPGD gene are inherited in an autosomal recessive manner, and the condition is sometimes abbreviated as PHOAR1 or Touraine-Solente-Gole syndrome.
Overview of pachydermoperiostosis syndrome
The three main features of pachydermoperiostosis are enlarged fingertips (clubbing), thickened facial skin (pachydermia), and excessive sweating (hyperhidrosis). Symptoms of the syndrome usually begin in childhood or adolescence, often around puberty, and progress slowly over about ten years [1].
Pachydermoperiostosis is the complete form of PHO in which there is also abnormal skin thickness (pachydermia). There are also two incomplete forms, one with bone problems with isolated skin changes, and another form characterized by pachydermia with minimal or no periosteal new bone growth (Figure 1).
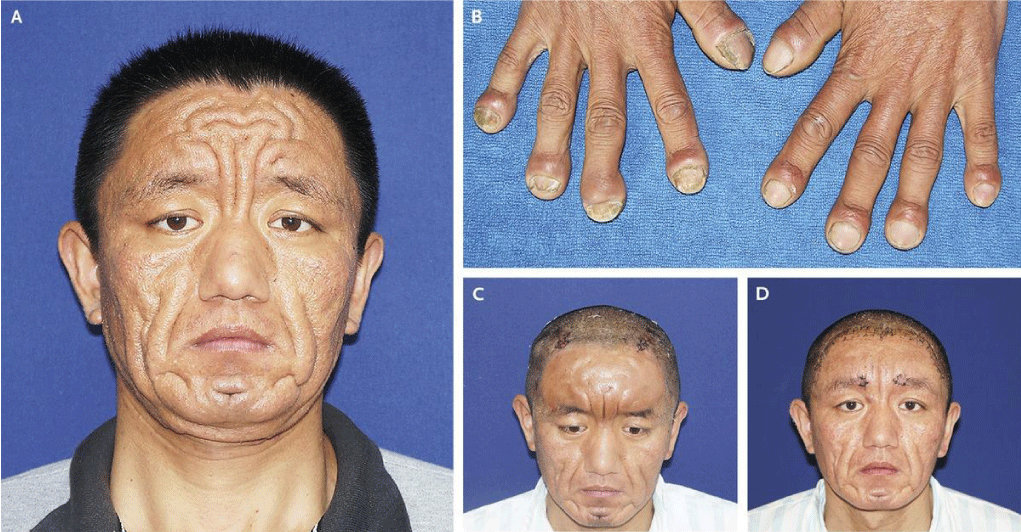
Figure 1: Clinical features of Pachydermoperiostosis syndrome showing skin involvement [1].
Clinical signs and symptoms of pachy-dermoperiosteal syndrome
PHO involves coarse facial features with oily, thick, grooved skin, joint pain, enlarged fingertips and toes, and hyperhidrosis of the hands and feet. Genetic mutations, particularly in the SLCO2A1 gene, contribute to this condition and may follow autosomal dominant or recessive inheritance patterns. Distinguishing PHO from similar conditions like rheumatoid arthritis or psoriatic arthritis is crucial for accurate diagnosis. Management primarily focuses on symptom relief, employing NSAIDs, colchicine, or corticosteroids to address pain and inflammation, while surgical interventions like vagotomy may offer improvement in severe cases. New bone growth (periosteal reaction), often at the ends of long bones, causes joint pain. Excess skin is seen on the scalp, causing deep grooves or ridges (cutis verticis gyrate) [1,2] (Figure 2).
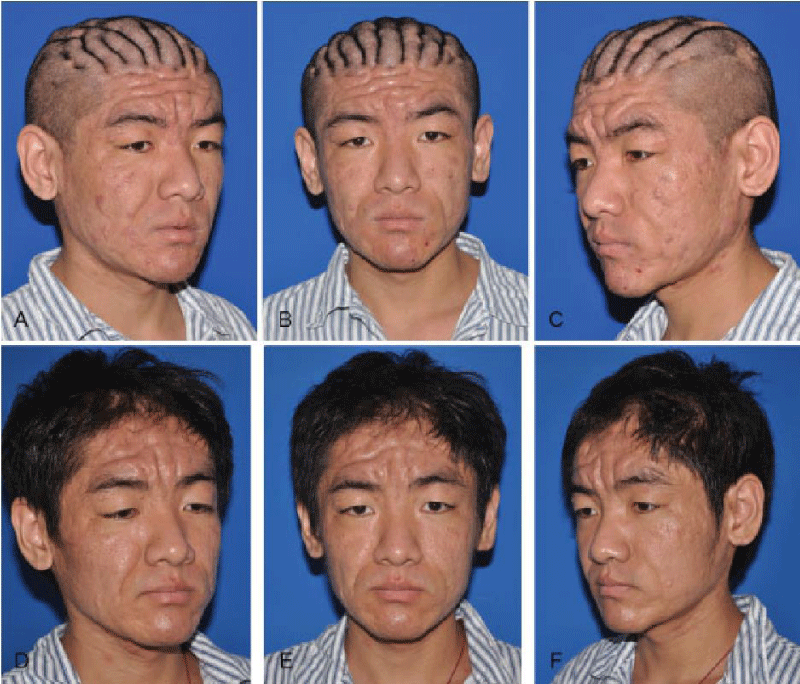
Figure 2: Clinical presentation of skin disorders in a patient diagnosed with PHO associated with pachydermoperiosteosis syndrome [1].
Other symptoms can include swelling or pain in large joints. Drooping eyelids (ptosis); a long-term skin condition that causes dry or moist, yellow scales (seborrheic dermatitis); ulcers; long eyelashes; and occasional diarrhea are some of the features. Other characteristics include swelling of hair follicles related to large open pores, heart disease at birth, or delayed closure of the space between the bones of the skull (fontanel). Symptoms vary from person to person, but in general, men tend to have more serious symptoms than women. X-rays can help check for features that are not noticeable to the naked eye [1,3] (Figure 3).
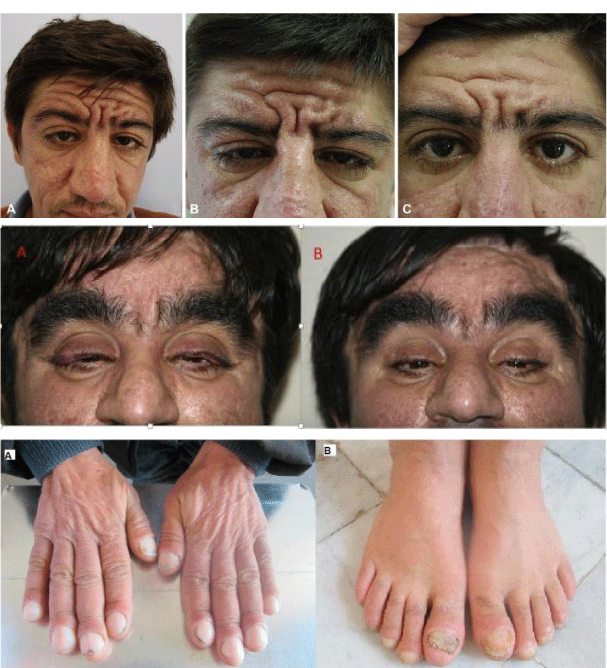
Figure 3: Another view of the types of disorders associated with pachydermoperiosteosis syndrome [1].
Etiology of pachydermoperiosteal syndrome
For most people, the diagnosis of PHO is based on clinical features. It is diagnosed more often in men than in women, as men tend to have more pronounced and severe features. PHO can be hereditary, but a non-genetic form has also been described. Genetic conditions can be inherited in different ways. Each individual typically inherits one allele of a gene from each parent. Genes provide instructions that tell the body how to function. When there is a harmful change (mutation) in a gene, it may not function normally, leading to unusual features or conditions. Autosomal dominant and autosomal recessive inheritance patterns have been reported for PHO [1,4].
Two genes are associated with PHO: the HPGD gene, located on the long arm of chromosome 4 at 4q34.1, and the SLCO2A1 gene, located on the long arm of chromosome 3 at 3q22.1 - q22.2. Mutations in the HPGD gene are inherited in an autosomal recessive manner, and the condition, sometimes abbreviated as PHOAR1, is also known as Touraine-Solente-Gole syndrome (HPGD mutations lead to Touraine-Solente-Gole syndrome) [1,5].
A recessive genetic condition occurs when a person inherits one inactive gene for the same condition from each parent. If a person receives one normal gene and one gene with a mutation, the person will be a carrier for the condition and will usually not show symptoms. The chance that two carrier parents will pass on both inactive genes and have a child with the condition is 25% with each pregnancy. The chance of having a child like the carrier parents with each pregnancy is 50%. The chance that a child will receive normal active genes from both parents is 25%. This chance is the same for both men and women. Parents who are closely related by blood (familial) have a higher chance than non-familial parents of both carrying the same inactive gene, which increases the chance of having children with a recessive genetic disorder [1,6] (Figure 4).
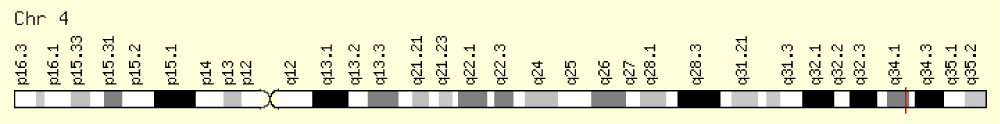
Figure 4: Schematic of the physical map of chromosome number 4, where the HGPD gene is located on the long arm of this chromosome as 4q34.1 [1].
PHO caused by mutations in the SLCO2A1 gene can be autosomal dominant or recessive. The autosomal recessive form is called PHOAR2. The autosomal dominant form is called PHOAD. Dominant genetic disorders occur when only one copy of an abnormal gene is needed to show symptoms of the disease. The inactive gene can be inherited from either parent or can be the result of a new mutation (gene change) in the affected person. The chance of passing the inactive gene from an affected parent to a child is 50 percent for each pregnancy. The risk is the same for men and women. However, with PHOAD, men are more commonly and often more severely affected [1,7] (Figures 5,6).
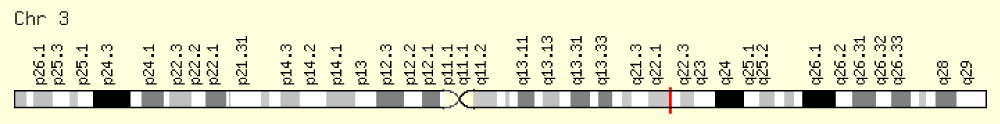
Figure 5: Schematic of the physical map of chromosome number 3, where the SLCO2A1 gene is located on the long arm of this chromosome as 3q22.1-q22.2 [1].
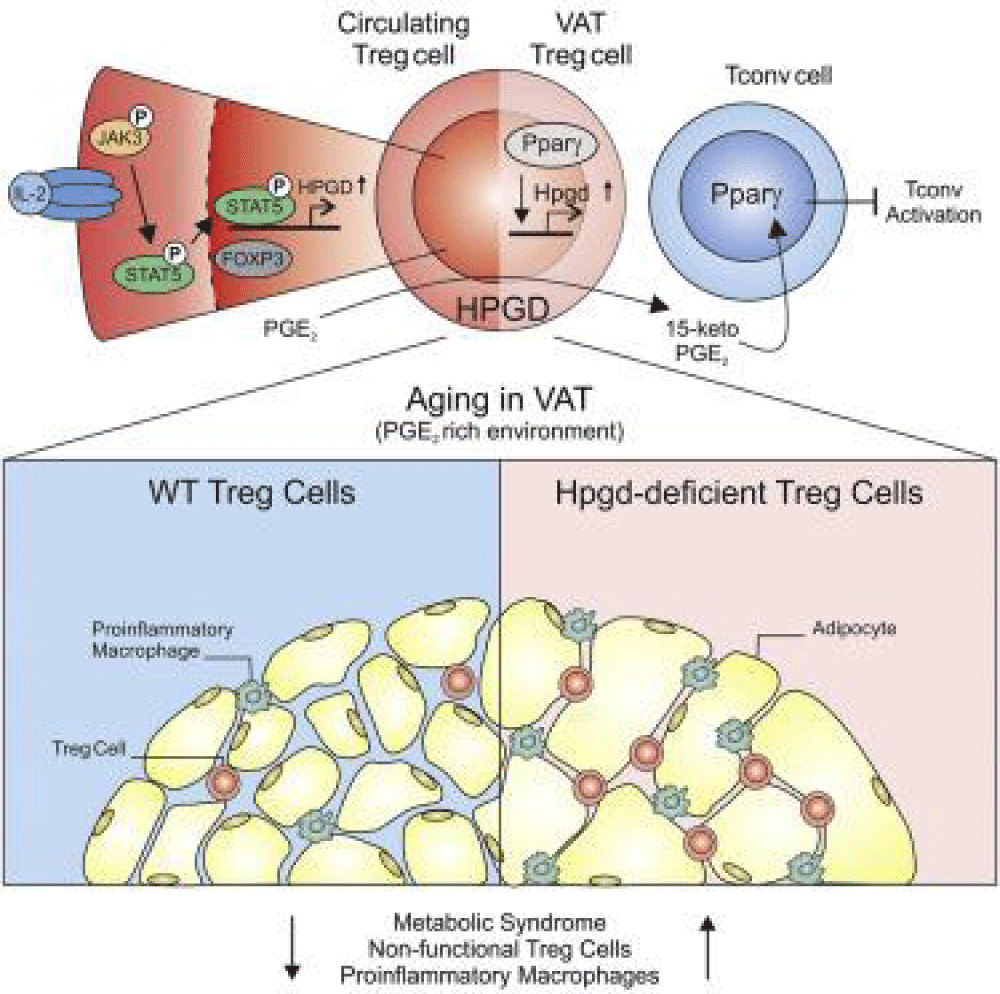
Figure 6: Role of HPGD in prostaglandin metabolism within adipocytes: Mechanism schematic [1].
Frequency of pachydermoperiosteal syndrome
PHO is a rare disorder that affects men more than women at a ratio of 7:1. This means that for every 7 men diagnosed with the condition, 1 woman will be diagnosed. Women, due to milder manifestations, are often underdiagnosed [1,8] (Figure 7).
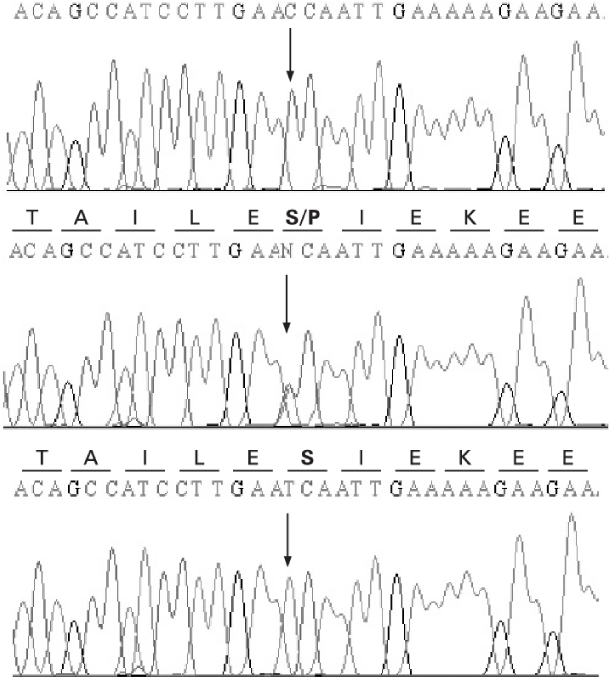
Figure 7: Schematic of a nucleotide mutation pattern in the HGPD gene [1].
Disorders associated with pachydermoperiosteal syndrome
The following disorders can have symptoms similar to those of pachydermoperiosteal syndrome. Comparing features can be helpful in making a correct diagnosis:
Acromegaly is a rare, gradually progressive, acquired disorder that affects adults. It is a condition in which excess growth hormone causes various tissues to grow larger than normal, causing the person to grow taller. The arms, legs, jaws, and face are most often affected. Enlargement of soft tissues, especially within the heart, is a serious feature of the disease [1,9] (Figure 8).
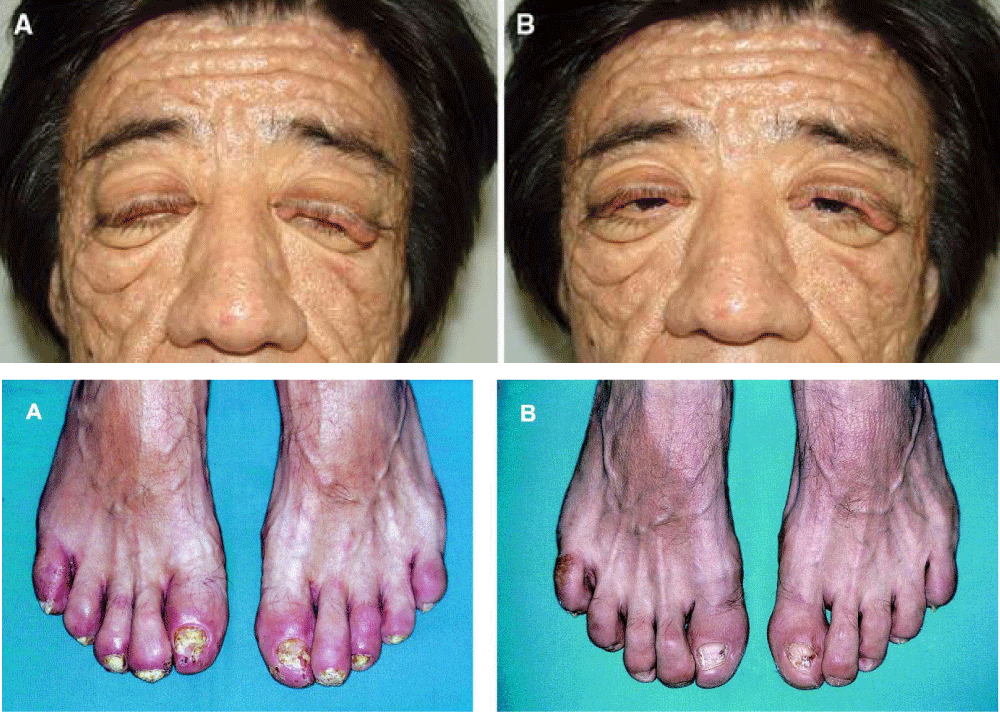
Figure 8: Illustrations of disorders associated with PHO syndrome [1].
Hypertrophic pulmonary osteoarthropathy (Bamberger-Marie disease) is a rare condition in which there is an abnormally large growth at the ends of the long bones, often the fingers and toes. It occurs in people with long-term lung disease, heart disease, and sometimes other disorders [1,10].
The features of PHO can be confused with osteoarthropathy (any condition that affects the bones and joints), psoriatic arthritis (arthritis associated with psoriasis - red and white patches on the skin), rheumatoid arthritis (a condition that can cause joint damage), and thyroid acropachy (thickening and swelling of the hands and feet) [1,11] (Figure 9).
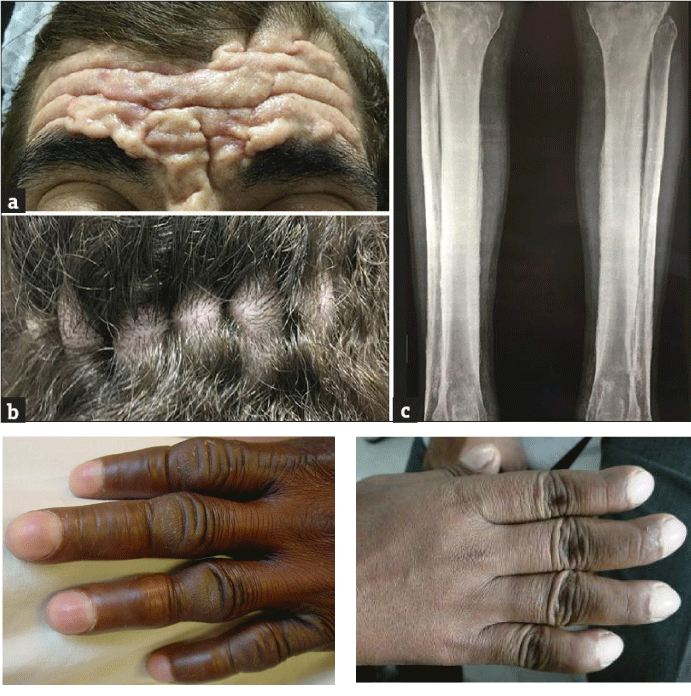
Figure 9: Images of skin and bone disorders associated with PHO syndrome [1].
Diagnosis of pachydermoperiosteal syndrome
The diagnosis of PHO is based on clinical features, although genetic testing is essential for definitive confirmation. The main diagnostic criteria are thickening of the fingers and new bone growth (periosteum) [1,12] (Figure 10).
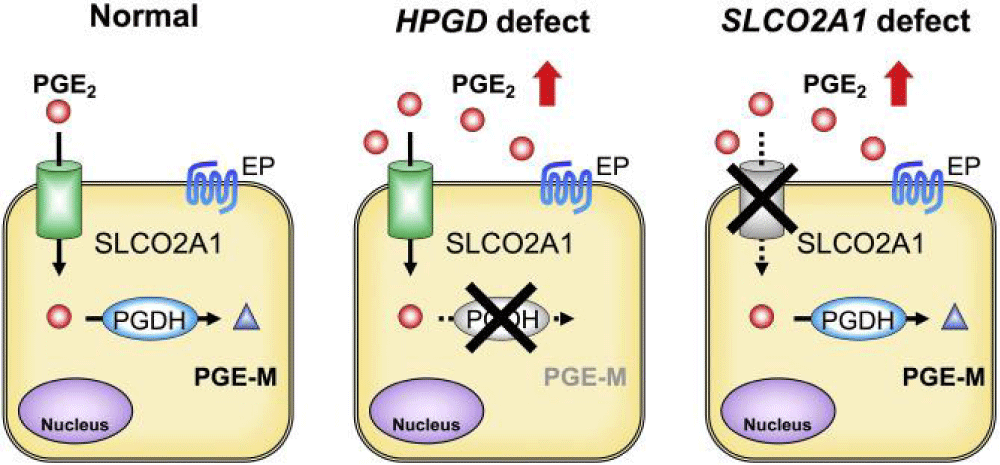
Figure 10: Schematic of the molecular and biochemical mechanism of decreased expression of SLCO2A1 and HGPD genes versus normal function of these genes [1].
Treatment for pachydermoperiosteal syndrome
Treatment remains largely symptomatic and supportive, focusing on pain management and skin symptoms. Nonsteroidal anti-inflammatory drugs (NSAIDs, such as Advil), colchicine, or corticosteroids may be used to reduce bone and joint pain. In rare cases, vagotomy, a surgical procedure in which certain branches of the vagus nerve are cut, may improve joint pain and swelling. Retinoids may be used to treat symptoms involving the skin. Plastic surgery may improve facial features, and surgical intervention can reduce digital clubbing. Genetic counseling may be helpful for patients and their families [1,13] (Figures 11,12).
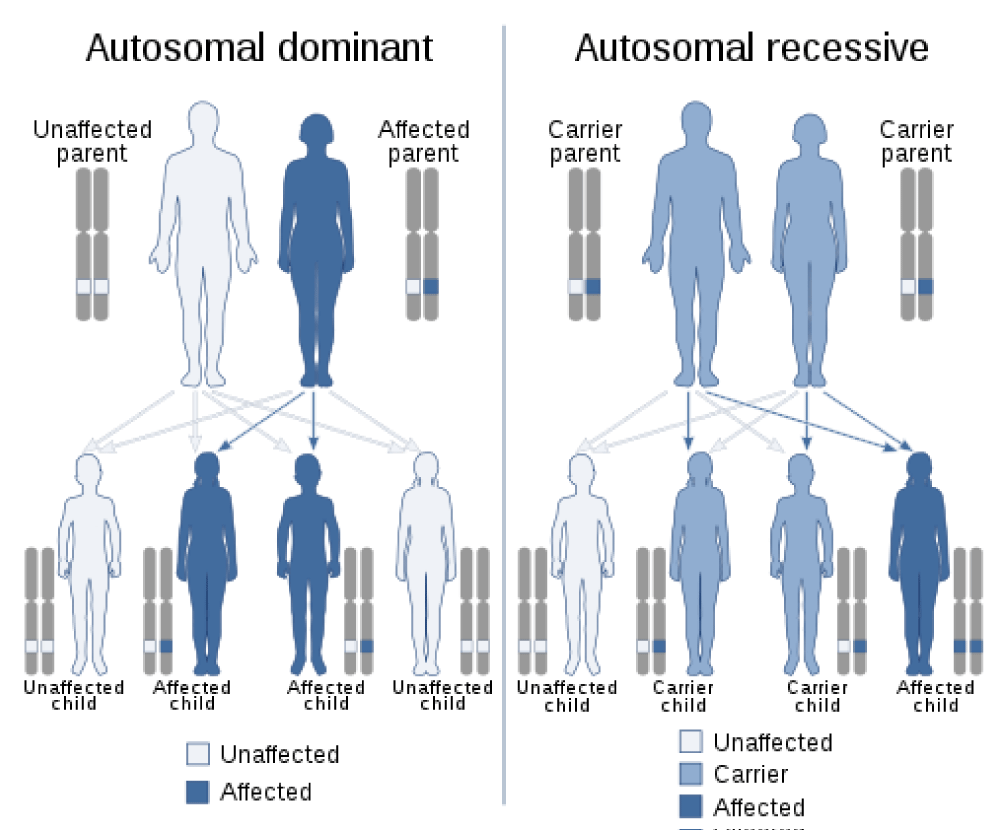
Figure 11: Schematic of the autosomal dominant (left) and autosomal recessive (right) inheritance patterns that PHO syndrome follows [1].
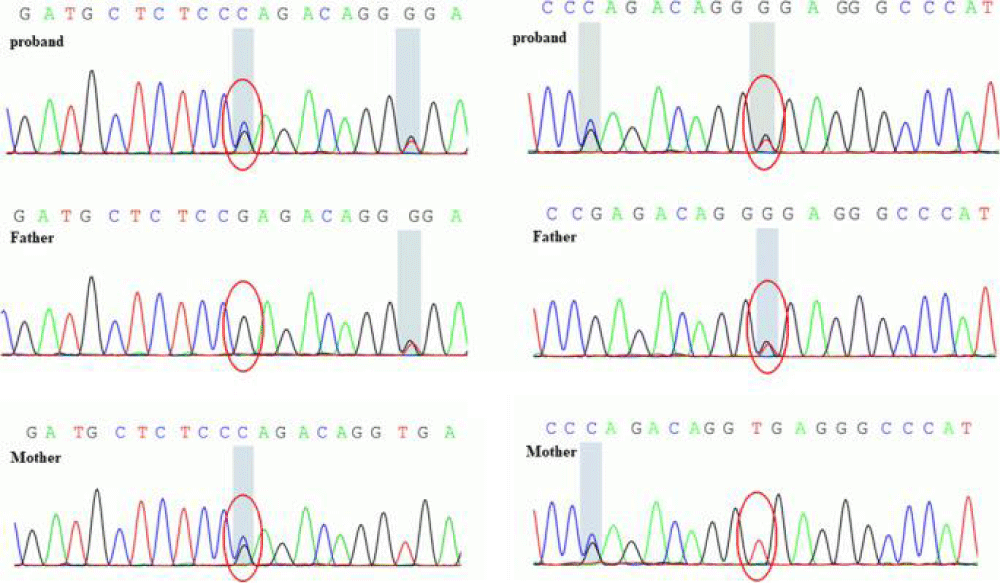
Figure 12: Schematic of a sample of nucleotide mutations in the SASLCO2A1 gene in parents and children with Pachydermoperiostosis Syndrome [1].
PHO is characterized by problems with skin and bone growth. Patients with PHO usually have coarse facial features with oily, thick, grooved skin on the face, joint pain, enlarged fingertips and toes, and excessive sweating of the hands and feet (hyperhidrosis). Genes provide instructions that tell the body how to function. When there is a harmful change (mutation) in a gene, it may not function normally, leading to unusual features or conditions. Autosomal dominant and autosomal recessive inheritance patterns have been reported for PHO. PHO caused by mutations in the SLCO2A1 gene can be autosomal dominant or recessive. The autosomal recessive form is called PHOAR2. The autosomal dominant form is called PHOAD [1,14]. In dominant inheritance, a single abnormal allele can cause disease manifestation. The features of PHO can be confused with osteoarthropathy (any condition that affects the bones and joints), psoriatic arthritis (arthritis associated with psoriasis - red and white patches on the skin), rheumatoid arthritis (a condition that can cause joint damage), and thyroid acropachy (thickening and swelling of the hands and feet). Treatment is mostly symptomatic and supportive. Nonsteroidal anti-inflammatory drugs (NSAIDs, such as Advil), colchicine, or corticosteroids may be used to reduce bone and joint pain. In rare cases, vagotomy, a surgical procedure in which certain branches of the vagus nerve are cut, may improve joint pain and swelling [1,15].
In summary, pachydermoperiostosis (PHO) is a genetically complex disorder, often involving mutations in the SLCO2A1 gene with autosomal dominant or recessive inheritance patterns. Precise genetic diagnosis is critical for distinguishing PHO from other conditions with overlapping features, such as rheumatoid arthritis and psoriatic arthritis. Clinical management focuses on alleviating symptoms, particularly bone and joint pain, through the use of NSAIDs, colchicine, or corticosteroids. In severe cases, surgical interventions like vagotomy may be considered. A multidisciplinary approach, including dermatological, rheumatological, and genetic expertise, is essential for effective care and improving quality of life for individuals with PHO (Figure 13).
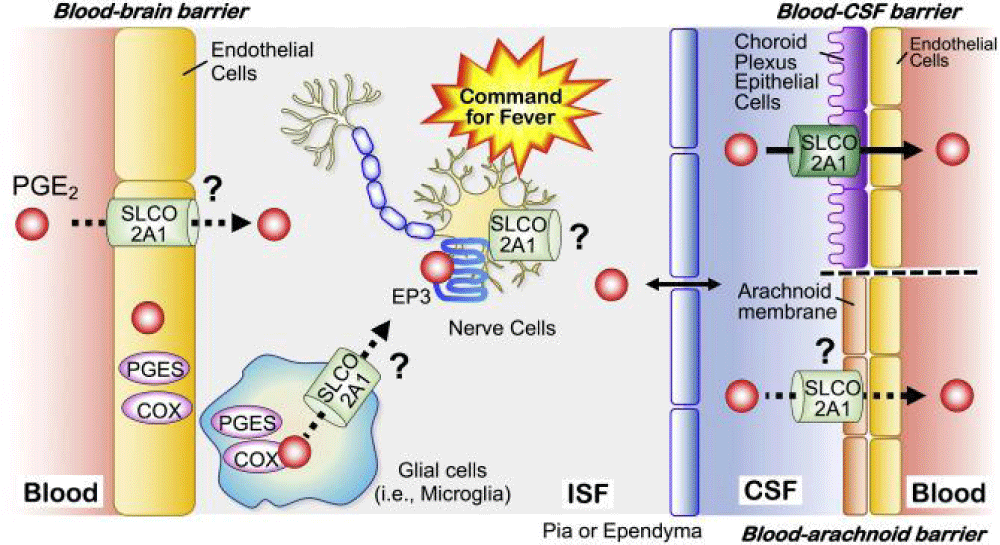
Figure 13: Schematic of the molecular and biochemical mechanism of the SLCO2A1 gene in nerve cells, cerebrospinal fluid, and blood vessels [1].
- Asadi S, Jamali M, Bagheri R, Dell SS. Pathology in Medical Genetic Books. Vol. 21 & 22. Iran: Amidi Publications; 2025. Available from: https://www.researchgate.net/publication/316861684_PATHOLOGY_IN_MEDICAL_GENETICS_2_M-Z
- Fauci AS, Braunwald E, Isselbacher KJ. editors. Harrison’s Principles of Internal Medicine. 20th ed. New York (NY): McGraw-Hill Companies. 2018.
- Griffiths C, Barker J, Bieiker T. Rook’s Textbook of Dermatology. 9th ed. London (UK): Wiley Blackwell Scientific Publications. 2016.
- Xu Y, Zhang Z, Yue H, Li S, Zhang Z. Monoallelic mutations in SLCO2A1 cause autosomal dominant primary hypertrophic osteoarthropathy. J Bone Miner Res. 2021;36(8):1459-68. Available from: https://www.researchgate.net/publication/352569346
- Marques P, Stelmachowska-Banas M, Collier D, Wernig F, Korbonits M. Pachydermoperiostosis mimicking the acral abnormalities of acromegaly. Endocrine. 2020;67(2):499–500. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7033267/
- Joshi A, Nepal G, Shing YK, Panthi HP, Baral S. Pachydermoperiostosis (Touraine–Solente–Gole syndrome): a case report. J Med Case Reports. 2019;13:39. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6362496/
- Yuan L, Liao RX, Lin YY, Jiang Y, Wang O, Li M, et al. Safety and efficacy of cyclooxygenase-2 inhibition for treatment of primary hypertrophic osteoarthropathy: a single-arm intervention trial. J Orthop Translat. 2019;18:109-18. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6546478/
- Primary hypertrophic osteoarthropathy related gastrointestinal complication has distinctive clinical and pathological characteristics: two cases report and review of the literature. Orphanet J Rare Dis. 2019;14:297. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6828001/
- Yuan L, Chen X, Liu Z, Wu D, Lu J, Bao G, et al. Novel SLCO2A1 mutations cause gender-differentiated pachydermoperiostosis. Endocr Connect. 2018;7(11):116-28. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6231361/
- Li SS, He JW, Fu WZ, Liu YJ, Hu YQ, Zhang ZL. Clinical, biochemical, and genetic features of 41 Han Chinese families with primary hypertrophic osteoarthropathy, and their therapeutic response to etoricoxib: results from a six-month prospective clinical intervention. J Bone Miner Res. 2017;32(8):1659-66. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5539869/
- Lee S, Park SY, Kwon HJ, Lee CH, Kim OH, Rhee Y. Identification of the mutations in the prostaglandin transporter gene, SLCO2A1 and clinical characterization in Korean patients with pachydermoperiostosis. J Korean Med Sci. 2016;31(5):735-42. Available from: https://doi.org/10.3346/jkms.2016.31.5.735
- Zhang Z, He JW, Fu WZ, Zhang CQ, Zhang ZL. Mutations in the SLCO2A1 gene and primary hypertrophic osteoarthropathy: a clinical and biochemical characterization. J Clin Endocrinol Metab. 2013;98(5):E923-E933. Available from: https://doi.org/10.1210/jc.2012-3568
- Hamosh A, Scott AF, Amberger J, Valle D, McKusick VA. Online Mendelian Inheritance in Man (OMIM). Hum Mutat. 2000;15(1):57-61. Available from: https://doi.org/10.1002/(sici)1098-1004(200001)15:1%3C57::aid-humu12%3E3.0.co;2-g
- Pachydermoperiostosis. Medscape. 2021. Available from: https://emedicine.medscape.com/article/1075122-overview
- Pachydermoperiostosis. Orphanet. 2011. Available from: https://www.orpha.net/en/disease/detail/2796