
More Information
Submitted: May 13, 2026 | Accepted: May 26, 2026 | Published: May 27, 2026
Citation: Kochumon SP, Nujoom N, Jagadeesan P, Scaria V, Vasudevan DM, Soman KP, et al. One-time CRISPR Adenine Base Editing Intervention in SMA: From SMN2 Splice Correction to Motor Neuron Rescue. J Genet Med Gene Ther. 2026; 9(1): 1-6. Available from:
https://dx.doi.org/10.29328/journal.jgmgt.1001014
DOI: 10.29328/journal.jgmgt.1001014
Copyright license: © 2026 Kochumon SP, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Spinal Muscular Atrophy (SMA): SMN1; SMN2; Motor neuron degeneration; Exon 7 Splicing; Adenine Base Editing (ABE); CRISPR-Cas9; Gene editing; Gene therapy

One-time CRISPR Adenine Base Editing Intervention in SMA: From SMN2 Splice Correction to Motor Neuron Rescue
Sheena P Kochumon1, Najma Nujoom2, Prem Jagadeesan3, Vinod Scaria4, DM Vasudevan2, KP Soman3 and Cherupally Krishnan Krishnan Nair2*
1Department of Paediatrics, Amrita Institute of Medical Sciences and Research Centre, Amrita School of Medicine, Amrita Vishwa Vidyapeetham, Kochi, India
2Department of Health Science Research, Amrita Institute of Medical Sciences and Research Centre, Amrita School of Medicine, Amrita Vishwa Vidyapeetham, Kochi, India
3Amrita School of Artificial Intelligence, Amrita Vishwa Vidyapeetham, Coimbatore, India
4Chief Data Officer, Karkinos Healthcare Pvt. Ltd., IBC Knowledge Park, Bhavani Nagar, Bengaluru, Karnataka, India
*Corresponding author: Cherupally Krishnan Krishnan Nair, Department of Health Science Research, Amrita Institute of Medical Sciences and Research Centre, Amrita School of Medicine, Amrita Vishwa Vidyapeetham, Kochi, India, Email: [email protected]
Spinal muscular atrophy (SMA) is a devastating autosomal recessive neuromuscular disorder characterized by progressive muscle weakness, atrophy, and respiratory failure due to selective degeneration of lower motor neurons arising from homozygous deletion of exon 7 (95%) or mutation in the SMN 1 gene (5%),with severity correlating with SMN2 copy number—from fatal Type1 to milder Type 4—affecting 1:6,000–10,000 births worldwide and burdening India with 1,500–2,000 annual cases amid diagnostic delays. Although the backup SMN2 gene compensates a bit for SMN deficiency, a critical C→T transition in exon 7 leads to exon skipping and production of a truncated, unstable and nonfunctional SMN protein. Recent advances in disease-modifying therapies-including antisense oligonucleotides, small-molecule splicing modifiers, and gene replacement-have significantly improved clinical outcomes; however, they do not restore endogenous SMN expression in all tissues and often require repeated administration. Despite these medications like Spinraza injections, Zolgensma gene therapy, Evrysdi pills that increase SMN protein, the condition still has got significant morbidity: Type 1 babies frequently die before the age of two, 60–95% develop scoliosis, which makes spinal injections uncomfortable and dangerous, and lifetime expenses for each patient surpass $2 million. What if we could edit the nucleotide base of SMN2(T6C) using ABE10 to make it emulate like SMN1 gene to restore stable functional SMN protein that would be the permanent cure for SMA. This cutting edge molecular tool “AI-based Adenine Base Editors” would facilitate an endogenous regulation, laying the groundwork for precision medicine in rare disease management.
Spinal muscular atrophy (SMA) is a severe, life-limiting inherited neuromuscular disorder characterized by progressive degeneration of lower motor neurons, resulting in muscle weakness, hypotonia, and, in its most aggressive forms, early mortality. Affecting approximately 1 in 6,000–10,000 live births worldwide, SMA remains one of the most common autosomal recessive genetic causes of infant death1. The disease arises primarily from mutations or deletions in the SMN1 gene located on chromosome 5q13, leading to insufficient production of survival motor neuron (SMN) protein, a critical factor for motor neuron maintenance and function. Disease severity is strongly influenced by the copy number of the nearly identical SMN2 gene, which partially compensates for SMN deficiency but produces limited amounts of functional protein [1].
Clinically, SMA presents as a spectrum ranging from prenatal-onset forms with profound weakness and respiratory failure to adult-onset phenotypes with relatively mild proximal muscle involvement. Historically, management was largely supportive, focusing on respiratory assistance, nutritional optimization, orthopedic care, and psychosocial support. However, the therapeutic landscape has changed dramatically in the past decade. The introduction of disease-modifying therapies—including antisense oligonucleotide treatment, small-molecule splicing modifiers, and adeno-associated virus–mediated gene replacement—has shifted SMA from a uniformly fatal pediatric condition to a treatable genetic disorder. Early intervention, particularly in presymptomatic infants identified through newborn screening, has demonstrated the greatest functional benefit, emphasizing the importance of early genetic diagnosis and population-level carrier screening strategies. Despite these advances, current treatments do not fully restore physiological SMN levels in all tissues, and long-term outcomes continue to be evaluated. Emerging approaches such as in utero gene therapy and precision genome editing—including CRISPR-based and base-editing platforms—offer the possibility of directly correcting pathogenic variants and achieving durable restoration of SMN expression. These innovations represent a conceptual shift from symptom modification toward molecular cure. As therapeutic capabilities expand, parallel progress in genetic counseling, carrier detection, and prenatal diagnostics remains essential for informed reproductive decision-making and early therapeutic access [1].
All individuals with SMA retain at least one copy of the SMN2 gene, which is nearly identical to SMN1 but differs by a critical single-base substitution (C·G to T·A) in exon 7. This seemingly minor C-to-T mutation disrupts normal RNA splicing, causing most SMN2 transcripts to skip exon 7 and produce a truncated, unstable, and largely non-functional protein. Consequently, only a small fraction of full-length, functional SMN protein is generated. Importantly, the number of SMN2 gene copies modifies disease severity, as additional copies can produce slightly higher levels of functional protein and are generally associated with a milder clinical phenotype.
Adenine base editing (ABE) offers a promising therapeutic strategy for SMA by permanently correcting the pathogenic nucleotide in SMN2. The goal is to convert the thymine (T) back to cytosine (C), effectively transforming SMN2 into a functional SMN1-like gene. Mechanistically, the ABE enzyme performs an A-to-G conversion on the DNA strand complementary to the mutated T, resulting in restoration of a stable G·C base pair and correction of the exon 7 splicing defect. Unlike traditional CRISPR nucleases, ABE does not introduce double-strand DNA breaks, thereby reducing the risk of unintended insertions or deletions. By directly modifying the endogenous SMN2 gene, this approach aims to provide a durable, potentially one-time treatment that restores physiologic SMN protein expression under native regulatory control, potentially eliminating the need for repeated dosing and reducing risks associated with protein overexpression seen in some current therapies.
The discovery of CRISPR/Cas9 (Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-associated protein 9) genome-editing technique offers new hope for the treatment of inherited genetic diseases by enabling precise, programmable modification of disease-causing DNA sequences in living cells [2]. This is made possible by an RNA-guided Cas9 endonuclease that allows DNA cleavage at any genomic locus [3]. However, conventional CRISPR-Cas9 editing relies on the need for a double-strand break (DSBs), which is predominantly repaired by the error-prone, non-homologous end joining (NHEJ) pathway [4]. The NHEJ pathway often results in unpredictable insertions or deletions (indels) of sequences at the target region, making the editing a difficult-to-control process. This process can occur at the target loci as well as at off-target sequences, making them a risky tool for genome editing [5]. The other DNA repair pathway, homology-directed repair (HDR), offers precise gene editing; its efficiency is very low in most of the cell types and is largely restricted to dividing cells [2,6]. It also requires the delivery of the donor template to the target cells. Together, these challenges make the use of HDR-based gene editing difficult to use for therapeutic applicability.
Importantly, the majority of the human genetic diseases, estimated to be over 50%, are caused by single-nucleotide variants (SNVs) [7]. Correcting such SNPs does not require the precise base-pair corrections. DSB-based strategies can be potentially unsafe for this purpose, as they can lead to chromosomal rearrangements, deletion of small to large portions, and even genomic instability [8].
These limitations pointed to the need for a strong genome-editing approach without making DSBs or the need for a donor DNA template, but at the same time, that could directly rewrite individual DNA bases to correct pathogenic point mutations. Base editing emerged as a precise mechanism to address this unmet need in genome therapeutics [9].
Discovery of base editing
Two independent biological insights have laid the foundation for the development of base editors (BEs) [9,10]. First, catalytically inactive (dCas9) or nickase version of Cas9 (Cas9n) could act as target-specific DNA-binding proteins capable of recruiting diverse effector proteins to specific genomic locations based on the guide RNA sequence complementarity [11]. Second, cytidine and adenine deaminases were known to catalyze specific base conversions in single-stranded nucleic acids during natural biological processes [10]. The combination of these two, the DNA-targeting Cas9 and a catalytic domain capable of deamination of cytidine or adenine base, resulted in chimeric proteins called base editors. BEs do not require DSBs for base modifications, thereby avoiding the risks associated with the generation of insertions or deletions at the on- and off- target sites. Different base editing systems are being reported so far by different groups, further expanding their targeting scope and specificity [9,12].
In 2016, Komor and colleagues reported the first cytosine base editor (BE1) by fusing a cytidine deaminase (APOBEC1) with a catalytically inactive Cas9 (dCas9) [13]. This combined system demonstrated targeted conversion of cytosine to uracil within a defined window of single-stranded DNA exposed during Cas9-mediated R-loop formation. Subsequent DNA replication or repair step replaced uracil with thymine, resulting in a permanent correction of C → T (or G → A) substitution. This demonstrated that precise base conversions are possible without inducing DSBs, opened a fundamental possibilities of targeted genome editing strategies. However, the first generation cytosine base editors exhibited modest efficiency.
In 2017, Gaudelli and his colleagues introduced adenine base editors (ABEs) significantly expanding the scope of base editing beyond just the cytosine conversions [10]. Unlike the cytosine base editors, ABEs rely on adenine deamination, a reaction for which no naturally occurring DNA adenine deaminses were known. To address this, the authors engineered a tRNA adenosine deaminase (TadA) from Escherichia coli through directed evolution, capable of acting on single-stranded DNA exposed during Cas9 mediated R-loop formation. This innovation broadened the applicability of base editing where ABEs enabled targeted A → G (or T → C) conversions. Together, these discoveries established based editing as a versatile platform for correction of the two most common transition mutations in the human genome.
Molecular mechanism of adenine deaminase based DNA base editing
Adenine base editors (ABEs) catalyze the deamination of adenosine to inosine within genomic DNA [10]. Inosine base pair with cytidine and is interpreted as guanine by cellular DNA polymerase during replication or DNA repair. Consequently, this process results in a permanent convertion of A into G or A-T into G-C base pairing at the target site [10].
To achieve this, adenine base editors are engineered as fusion proteins that combine a programmable DNA binding protein with a catalytic domain of adenine deaminases. For this application, ABEs requires three essential components: (i) a programmable DNA-targeting module, typically a Cas9 nickase (Cas9n) or catalytically inactive Cas9 (dCas9), directed to the target sequence by a single-guide RNA (sgRNA); (ii) a adenine deaminase domain, capable of catalyzing adenine deamination on single-stranded DNA; and (iii) auxiliary domains that modulate endogenous DNA repair pathways and bias the cell toward the retention of the edited base [1].
Upon binding to the target locus, the Cas9 /sgRNA complex forms an R-loop, which displaces one DNA strand and exposes a short stretch of single-stranded DNA [14]. Within this exposed regions, known as the editing window, the tethered deaminase could catalyse the base conversion. Cytosine deaminase convert cytosine to uracil, while adenine deaminase convert adenine to inosine, which is read as guanine in the DNA replication or DNA repair process. Both of these conversions take place without the need of DNA double strand breaks [9,10].
When a Cas9 nickase (Cas9n) enzyme is used in this process, they introduces a single-strand break in the non-edited strand, which directs the mismatch repair mechanism to replace the unedited base using edited strand as the template [11]. This strategic manipulation of endogenous repair pathway allows efficient and predictable base substitution without the need for DSB-associated genomic instability. The efficiency and specificity of base editing are influenced by multiple parameters, including the width of editing window, local sequence context surrounding the target sequence, the availability of the protospacer adjacent motif (PAM), chromatin state, and cellular DNA repair activity and its efficiency. Additionally, strand orientation, guide RNA design, and cell type-specific repair dynamics further contribute to variability in editing outcomes [15].
PAM constraints and evolution of base editors
The availability of compatible protospacer adjacent motif (PAM) is an important criteria for the Cas9 binding and target recognition [3]. First-generation base editors predominantly employed Streptococcus pyogenes Cas9 (SpCas9), which recognizes an NGG PAM sequence located immediately upstream of the guide RNA binding sequence. This NGG PAM sequence requirement also restricts target accessibility and limits the potential to edit many disease-relevant pathogenic mutations that do not lie proximal to an NGG motif.
To overcome this constraints, extensive protein engineering was employed for the development 0f Cas9 variants with expanded or relaxed PAM specificities, thereby to significantly increase the editable fraction of the genome [9]. In parallel, efforts were focused on optimizing the deaminase domain o improve its catalytic activity, and specificity. In the case of ABEs, successive rounds of protein engineering yielded highly active variants, including ABE7.10, which demonstrated editing efficiencies exceeding 50% within an optimal editing window spanning positions 4 – 7 relative to the PAM [16]. Further generations of ABEs (e.g., ABE6.3, ABE7.8, and later ABE8 variants) were developed to further enhance catalytic efficiency, broaden sequence compatibility, and reduce off-target activity [17,18]. A key advantage of ABEs is that inosine repair is inefficient in eukaryotic cells, allowing the edited base to persist and be faithfully converted to guanine during DNA replication.
In 2020, a group of researchers including Jennifer Doudna, David R. Liu, and their collaborators reported the development of ABE8e, derived from ABE7.10 using phage – assisted non-continuous and continuous evolution (PANCE and PACE). ABE8e includes eight additional mutations, and demonstrated up to 590- fold increase in catalytic activity compared to earlier ABE varients. Most importantly, enhanced editing efficiency were reported when ABE8e was combined with diverse Cas9 and Cas12 homologs, further expanding the scope and versatility of adenine base editing [19,20].
Applications of ABEs in the Treatment of Spinal Muscular Atrophy
Genome editing of SMN2 gene to restore the physiologically normal levels of SMN protein has become a major focus with advancements in CRISRP/cas9 based base-editing technology [19]. A major therapeutic concept in SMA treatment is to permanently correct the functional difference between SMN1 and SMN2 by editing the critical nucleotide in SMN2 exon7 (C6>T) back toward the SMN-1 like state. This correction promotes exon 7 inclusion during the splicing steps and increases the production of full-length SMN protein.
In 2023, A team of scientist led by David R. Liu demonstrated the therapeutic potential of this strategy using ABEs to replace single thymine (T) nucleotide in SMN2 exon 7 with cytosine (C), thereby functionally converting the SMN2 into and SMN1-like gene (Figure 1) [19]. This approach successfully restored SMN2 restored the SMN transcript and protein levels to that of wild type cells (~40 fold increase) in patient derived cells. This method also ensured minimal off-target effect across the genome. In vivo, intracerebroventricular injection of AAV9 vector encoding ABE showcased 87% conversion of SMN2 C6T in the transducted cells of the severe SMA∆7 mice models ( Smn -/- carrying human SMN2 +/+ and SMN∆7). Notably, this genome-editing approach resulted in a marked increase in life span, which was an average of 111 days when AAV-ABE was combined with Nusinersen, compared to the 28 days reported with Nusinersen alone treatment. Overall, AAV-ABEs-treated mouse showed a 6.5 times longer survival rate than the untreated animals, highlighting the therapeutic impact of SMN2 correction using base editors [19,20].
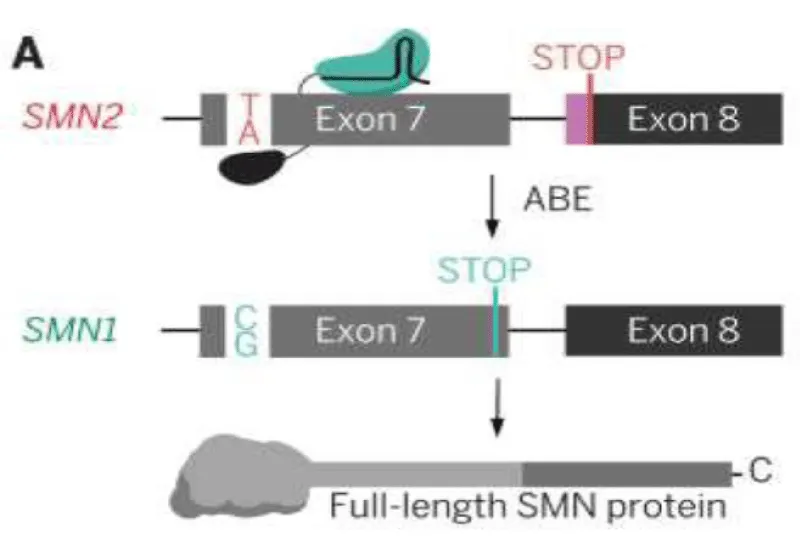
Figure 1: Diagram showing the conversion of A → G by ABEs at the Exon 7 C6T position. This conversion convert the SMN2 into a SMN1 phenotype to produce full length SMN protein.
Further optimization of this strategy was reported by Alves C, et al., which was systematically evaluated more than 100 combinations of guide RNAs (gRNAs) and ABEs to identify the most suitable combinations to correct SMN2 C6T mutations [21]. Using a near-PAMless CRISPR-Cas9 enzyme, named SpRY [5], they demonstrated upto 99% precise editing in SMA patient-derived fibroblasts with no detectable bystander editing, underscoring the importance of PAM flexibility and editor selection for maximizing therapeutic precision [17].
Another powerful therapeutic approach is to edit intronic splicing silencers that repress exon 7 inclusion in SMN2, especially in ISS-N1, a clinically validated regulatory element. ABE systems using broad-PAM Cas variants (E.g., SpRY) have been used to place edits into splicing regulatory motifs such as IIS-N1, driving increased exon 7 inclusion and SMN expression [19]. This strategy is particularly attractive because it targets the regulatory architecture controlling splicing rather than only one exonic nucleotide.
Multiple preclinical studies have demonstrated that precise base editing of regulatory nucleotides within SMN2 exon 7 or its surrounding splicing control elements can shift towards exon 7 inclusion, thereby increasing full length SMN protein as illustrated in Figure 1. Collectively, these studies demonstrate that adenine base editing enables efficient, durable, and highly specific correction of SMN2, providing strong proof-of-concept for its further development toward clinical application in SMA.
Despite promising editing outcomes, efficient delivery of ABEs to motor neurons and spinal cord tissue remains a major bottleneck. The large size of ABEs complicates its packaging into a single adeno-associated virus (AAV) vector, often necessitating dual-AAV strategies or alternative delivery platforms, which can reduce editing efficiency and increase variability across tissues.
Compared to conventional CRISPR/Cas9 nuclease-based strategies, base editing approaches showed minimal indel formation and reduced genomic disruption, an important consideration for motor neurons and other post-mitotic cells affected in SMA. This reinforced the suitability of ABEs for neuromuscular disorders where genomic integrity is critical.
Despite these advantages, most of the preclinical studies reported partial editing efficiencies, resulting in mosaic correction of SMN2 rather than uniform modification across all target cells. Most notably, even modest increases in SMN can yield therapeutic benefit; however, this variability highlights the need for improved editor activity, optimized delivery timing, and better tissue-specific targeting, particularly within the central nervous system.
Furthermore, editing window of ABEs might include multiple adenines, introducing the possibility of bystander edits, particularly in regulatoy regions that control splicing. Additionally, concerns, regarding off-target DNA and RNA deamination persist, requiring high-fidelity ABE variants and comprehensive safety profiling before clinical transitions.
Artificial intelligence (AI) is increasingly shaping the future of gene editing by enabling data-driven optimization of precision therapeutics for monogenic disorders such as spinal muscular atrophy (SMA). Recent studies highlight how AI models can systematically integrate genomic, transcriptomic, and epigenomic data to improve target selection, guide RNA design, and prediction of editing efficiency and specificity in CRISPR-based systems [22]. In the context of base editing, machine learning frameworks are being used to predict editing windows, minimize bystander edits, evaluate PAM flexibility, and estimate off-target risks across diverse cellular environments [23]. These computational approaches significantly accelerate experimental design cycles and reduce trial-and-error optimization. Beyond sequence-level engineering, AI can support rational vector design, tissue-specific delivery optimization to motor neurons, and modeling of long-term therapeutic response. The integration of AI with adenine base editing platforms therefore represents a powerful translational strategy to enhance precision, safety, and clinical scalability in SMA gene correction efforts.
Editing the nucleotide base of SMN2(T6C) using ABE10 makes it emulate like SMN1 gene by restoring stable functional SMN protein as a permanent cure for SMA.
As base editing approaches showed minimal indel formation and reduced genomic disruption, ABE is better suited for SMA
Integration of AI with adenine base editing platforms therefore represents a powerful translational strategy to enhance precision, safety, and clinical scalability in SMA gene correction efforts.
Apart from sequence-level engineering, AI can support rational vector design, tissue-specific delivery optimization to motor neurons, and long term therapeutic response.
Conflicts of Interest: The authors declare no conflict of interest.
Sources of funding and support: The authors received no funding related to this article.
Author contributions statement: All authors contributed equally.
Transfer the copy right: All authors agree to transfer the copy right to the journal
Competing interest policy: The authors declare no competing financial and/or non-financial interests concerning the article.
- Kochumon SP, Nair CKK. Management and Therapeutic Strategies for Spinal Muscular Atrophy. J Genet Med Gene Ther. 2024; 7: 001-007. Available from: https://dx.doi.org/10.29328/journal.jgmgt.1001009
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013 Nov;8(11):2281-2308. Available from: https://dx.doi.org/10.1038/nprot.2013.143
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012 Aug 17;337(6096):816-21. Available from: https://dx.doi.org/10.1126/science.1225829
- Mao Z, Bozzella M, Seluanov A, Gorbunova V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle. 2008 Sep 15;7(18):2902-6. Available from: https://dx.doi.org/10.4161/cc.7.18.6679
- Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181-211. Available from: https://dx.doi.org/10.1146/annurev.biochem.052308.093131.
- Lee K, Conboy M, Park HM, Jiang F, Kim HJ, Dewitt MA, Mackley VA, Chang K, Rao A, Skinner C, Shobha T, Mehdipour M, Liu H, Huang WC, Lan F, Bray NL, Li S, Corn JE, Kataoka K, Doudna JA, Conboy I, Murthy N. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat Biomed Eng. 2017;1:889-901. Available from: https://dx.doi.org/10.1038/s41551-017-0137-2.
- Eichler, E. E. Genetic Variation, Comparative Genomics, and the Diagnosis of Disease. N. Engl. J. Med. 381, 64 (2019). Available from: https://dx.doi.org/10.1056/NEJMra1809315
- Kosicki M, Tomberg K, Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat Biotechnol. 2018 Sep;36(8):765-771. Available from: https://dx.doi.org/10.1038/nbt.4192.
- Porto EM, Komor AC, Slaymaker IM, Yeo GW. Base editing: advances and therapeutic opportunities. Nat Rev Drug Discov. 2020 Dec;19(12):839-859. Available from: https://dx.doi.org/10.1038/s41573-020-0084-6
- Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, Liu DR. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature. 2017 Nov 23;551(7681):464-471. doi: 10.1038/nature24644. Epub 2017 Oct 25. Erratum in: Nature. 2018 Jul;559(7714):E8. Available from: https://dx.doi.org/10.1038/s41586-018-0070-x.
- Eid A, Alshareef S, Mahfouz MM. CRISPR base editors: genome editing without double-stranded breaks. Biochem J. 2018 Jun 11;475(11):1955-1964. Available from: https://dx.doi.org/10.1042/BCJ20170793.
- Bhatia P, Sterneckert J. Base editing: a promising tool to rescue spinal muscular atrophy. Signal Transduct Target Ther. 2023 Sep 25;8(1):362. Available from: https://dx.doi.org/10.1038/s41392-023-01583-5.
- Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016 May 19;533(7603):420-4. Available from: https://dx.doi.org/10.1038/nature17946.
- Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014 Mar 6;507(7490):62-7. Available from: https://dx.doi.org/10.1038/nature13011.
- Li, Z. et al. Engineering of high-precision C-to-G base editors with expanded site selectivity and target compatibility. Nucleic Acids Res. 53, (2025). Available from: https://dx.doi.org/10.1093/nar/gkaf717
- Gaudelli NM, Lam DK, Rees HA, Solá-Esteves NM, Barrera LA, Born DA, Edwards A, Gehrke JM, Lee SJ, Liquori AJ, Murray R, Packer MS, Rinaldi C, Slaymaker IM, Yen J, Young LE, Ciaramella G. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat Biotechnol. 2020 Jul;38(7):892-900. Available from: https://dx.doi.org/10.1038/s41587-020-0491-6.
- Walton RT, Christie KA, Whittaker MN, Kleinstiver BP. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science. 2020 Apr 17;368(6488):290-296. Available from: https://dx.doi.org/10.1126/science.aba8853.
- Li, S., Xu, K., Li, G. et al. Engineering the MmeFz2-ωRNA system for efficient genome editing through an integrated computational-experimental framework. Nat Commun 17, 1867 (2026). Available from:https://doi.org/10.1038/s41467-026-68644-5
- Arbab M, Matuszek Z, Kray KM, Du A, Newby GA, Blatnik AJ, Raguram A, Richter MF, Zhao KT, Levy JM, Shen MW, Arnold WD, Wang D, Xie J, Gao G, Burghes AHM, Liu DR. Base editing rescue of spinal muscular atrophy in cells and in mice. Science. 2023 Apr 21;380(6642):eadg6518. Available from: https://dx.doi.org/10.1126/science.adg6518.
- Richter MF, Zhao KT, Eton E, Lapinaite A, Newby GA, Thuronyi BW, Wilson C, Koblan LW, Zeng J, Bauer DE, Doudna JA, Liu DR. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat Biotechnol. 2020 Jul;38(7):883-891. Available from: https://dx.doi.org/10.1038/s41587-020-0453-z.
- Alves CRR, Ha LL, Yaworski R, Lazzarotto CR, Christie KA, Reilly A, Beauvais A, Doll RM, de la Cruz D, Maguire CA, Swoboda KJ, Tsai SQ, Kothary R, Kleinstiver BP. Base editing as a genetic treatment for spinal muscular atrophy. bioRxiv [Preprint]. 2023 Jan 21:2023.01.20.524978. Available from: https://dx.doi.org/10.1101/2023.01.20.524978.
- Erdoğan S. Integration of Artificial Intelligence and Genome Editing System for Determining the Treatment of Genetic Disorders. Balkan Med J. 2024 Oct 31;41(6):419-420. Available from: https://dx.doi.org/10.4274/balkanmedj.galenos.2024.2024-080824.
- Pandey S, Choudhari JK, Tripathi A, Singh A, Antony A, Chouhan U. Artificial Intelligence-Based Genome Editing in CRISPR/Cas9 Methods Mol Biol. 2025;2952:273-282. Available from: https://dx.doi.org/10.1007/978-1-0716-4690-8_16.